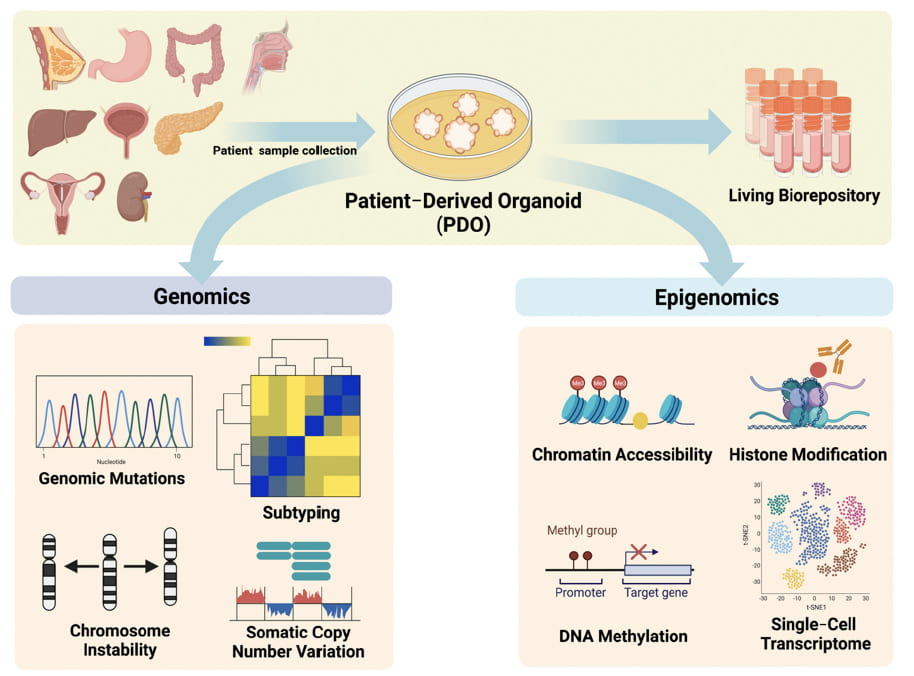
Genomic and Epigenomic Characterization of Tumor Organoid Models
Our group has previously established human 3D organoid model systems for different organs, such as Barret’s esophagus, gastroesophageal junction, oral cavity, among other tissues (Sci Tran Med 2022, Cancer Letter 2018, Nature Communication 2022). Recently, we have developed organoid models directly from normal squamous epithelium, dysplasia, pre-malignancy and SCC tumor samples, spanning the stepwise neoplastic evolution of SCC. Moreover, we have achieved robust success in reproducibly manipulating these structures for genomic editing by CRISPR/Cas9. We will utilize this novel model system to investigate genomic alterations and key pathways promoting SCC.
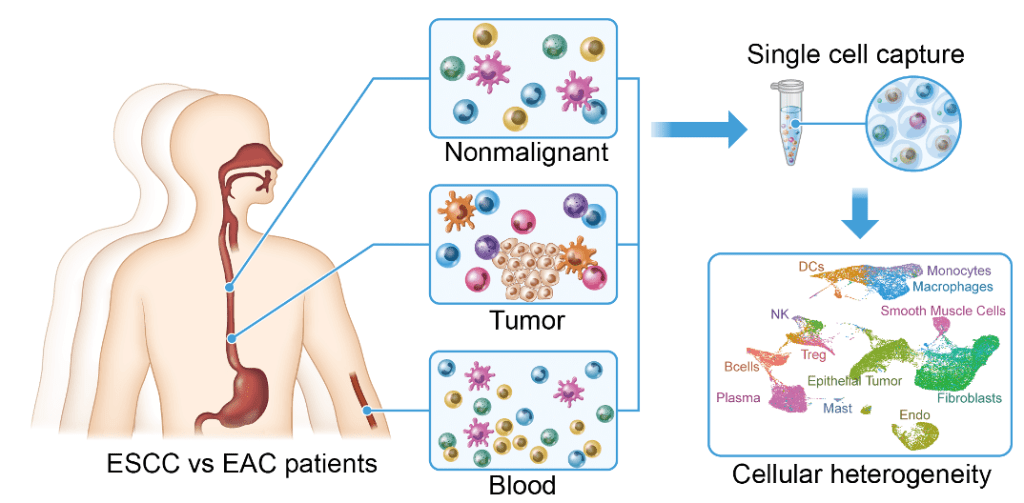
We are interested in the tumor microenvironment of GI cancers, and are using cutting edge genomic tools to dissect it in both animal models and patient samples, such as single-cell RNA analysis and single-cell ATAC-seq. We are studying the tumor immunity based on the findings from these single-cell genomics (Nature Communications 2021).
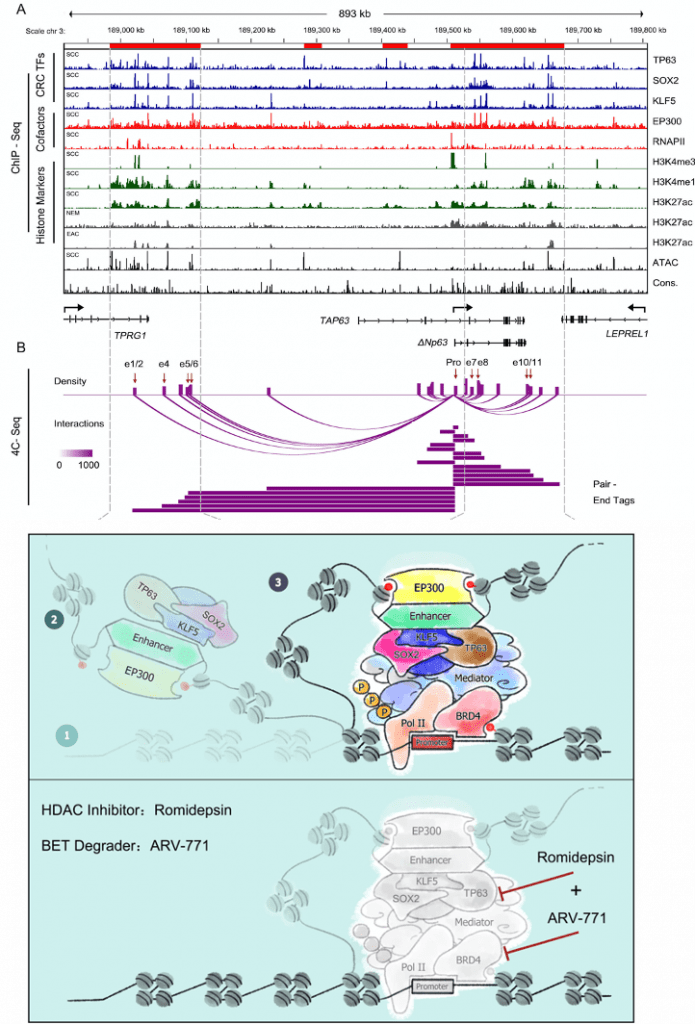
We have identified master regulator TFs using novel bioinformatic methods based on Chip-seq, ATAC-seq and RNA-seq data from both esophageal adenocarcinoma (Gut 2018; Gut 2019 ) and squamous cancer (Nature Communications 2018). We further revealed the epigenomic vulnerability of these GI tumors and tested the anti-tumor activity of BET and HDAC inhibitors (Gastroenterology 2020). We are continuing to study the epigenomic features of GI cancers for the development of more innovative targeting strategies.
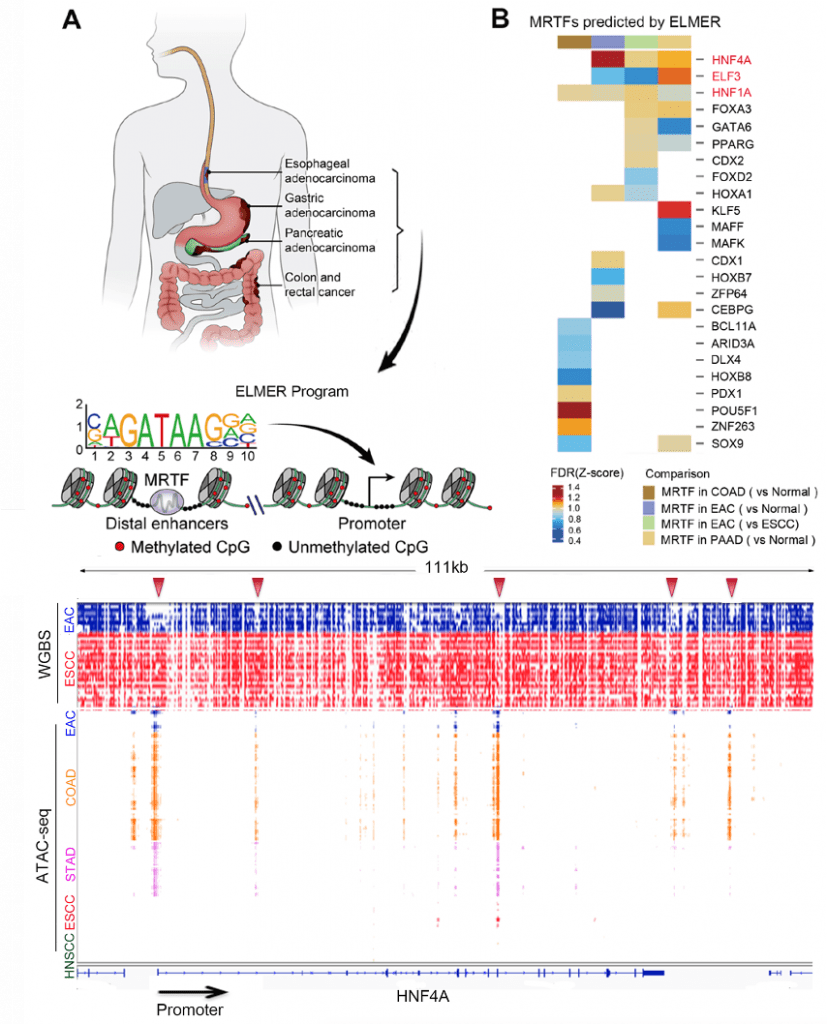
In collaboration with Ben Berman’s lab, we have recently developed a novel computational algorithm, ELMER, and identified cancer-specific master regulator TFs (such as HNF4A) from GI patient samples using DNA methylation profiles and matched RNA-seq data (Bioinformatics 2019; Cancer Res 2020).
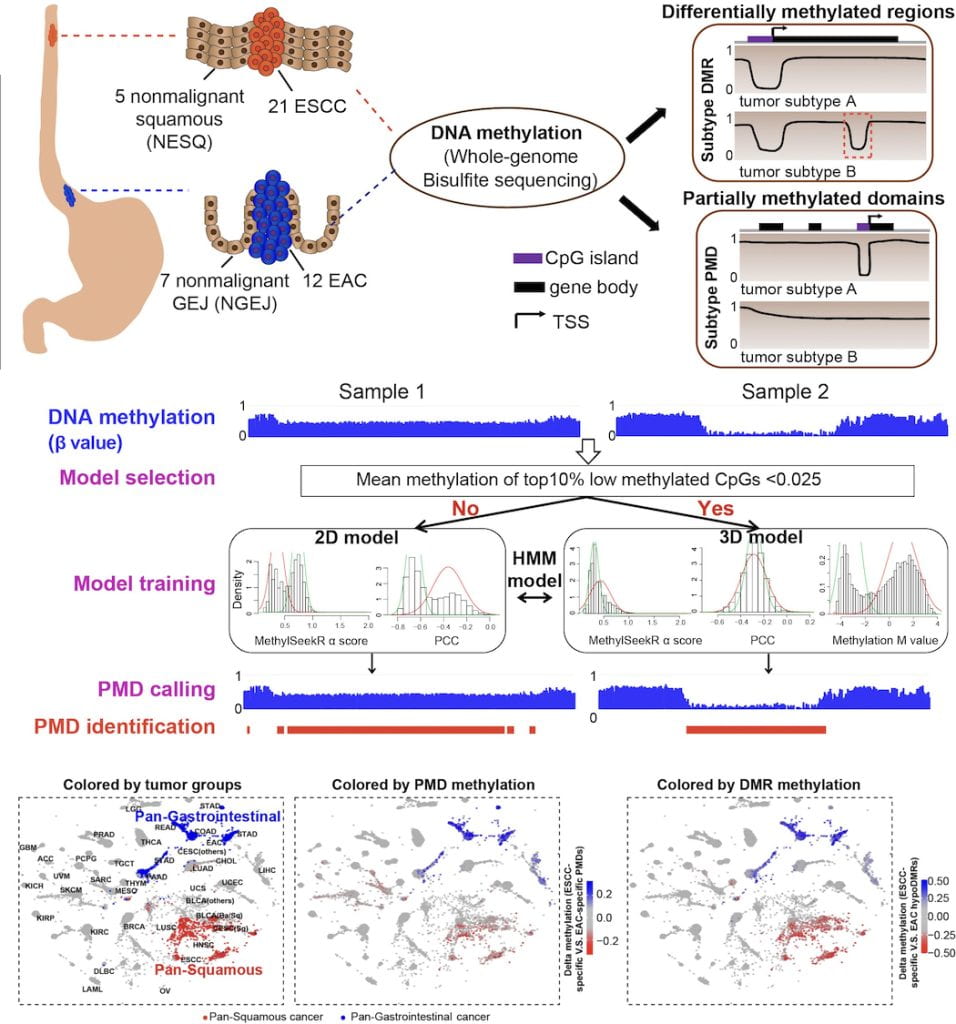
We develop a novel sequence-aware method to identify large partially methylated domains (PMDs), revealing profound heterogeneity at both the methylation level (depth) and genomic distribution (breadth) of PMDs across tumor samples. We find that cell-type-specific deposition of H3K36me2 may underlie the genomic distribution PMDs. At a smaller genomic scale, both cell-type- and cancer-specific differentially methylated regions (DMRs) are identified for each subtype. Using binding motif analysis within these DMRs, we show that a cell-type-specific transcription factor such as HNF4A can maintain the binding sites that it establishes in normal cells, while being recruited to new binding sites with novel partners such as FOSL1 in cancer. Finally, leveraging pan-tissue single-cell and pan-cancer epigenomic datasets, we demonstrate that a substantial fraction of the cell-type-specific PMDs and DMRs identified here in esophageal cancer, are actually markers that co-occur in other cancers originating from related cell types.