
Mayah Hijazi M.D, Wafaa Elatre M.D.


Mayah Hijazi M.D, Wafaa Elatre M.D.
Brian Ma, Wafaa Elatre
Clinical Summary:
56-year-old male with a history of sigmoid adenocarcinoma status-post resection and chemotherapy presents with new onset of intermittent hematuria for the past 5 months. On imaging, a large papillary mass in the posterior bladder wall and a colovesicular fistula is present. A cystoscopy was performed and a bladder washing is collected.
A ThinPrep slide and cell block is prepared. A cellular ThinPrep preparation shows several singly dispersed and groups of atypical cells with high nuclear to cytoplasmic ratio, anisonucleosis, irregular nuclear membranes, and hyperchromasia. Some are columnar. Admixed in the background are mixed inflammatory cells, erythrocytes, and bacterial colonies.
Cytology Description:
The cell block shows abundant atypical glands with large, pleomorphic, irregular nuclei, variably prominent nucleoli with moderate amounts of cytoplasm. The following immunohistochemistry was performed: CK20, CDX2, GATA3.
Given the history of sigmoid adenocarcinoma, positive immunostaining for CK20 and CDX2 with negative immunostaining for GATA3, the final diagnosis rendered is: Malignant cells present consistent with patient’s known history of colon primary.

Figure 1 (ThinPrep Slide

Figure 2 (Cell block)

Figure 3 (CDX2)

Figure 4 (CK20)

Figure 5 (GATA3)
Discussion: This case highlights the importance of history. Blindly examining this patient’s bladder washing could easily lead one to a diagnosis of high-grade urothelial carcinoma, however given the patient’s history of sigmoid adenocarcinoma, further workup was necessary. While a new primary high grade urothelial carcinoma is possible, the presence of a colovesicular fistula is concerning for extension of disease from colon. History and use of immunostains in this case helped confirm the final diagnosis
Joaquin Ponce-zepeda, Wafaa A. Elatre
69-year-old male was identified to have a 7.0 cm mass involving the body of the pancreas on CT Abdomen. Gastroenterology was consulted, and an Endoscopic Ultrasound (EUS) procedure was performed.
The EUS showed a large, heterogeneous, round mass in the pancreatic body with extension into the neck of the pancreas, measuring 4.0 cm by 3.0 cm. The borders of the mass were poorly defined by ultrasound. The remainder of the pancreas appeared atrophied as it was surrounded by adipose tissue.
Fine needle aspiration was performed with a 25 gauge needle using a trans-gastric approach.
The cytology specimen obtained during EUS consisted of 2 smears (Diff Quick preparations), and one cell block. Overall the smears and the cell block preparation contained exemplary cellularity, displaying multiple groups, cohesive clusters, and single acinar cells. The Diff Quick smears demonstrated a monomorphic acinar cell population with hyperchromatic nuclei predominantly in clusters.
The cell block displayed clusters of acinar cells with round nuclei, and abundant finely vacuolated to granular cytoplasm. Morphologically the smears and cell block constructed a picture of an acinic cell neoplasm (Figure 1, 2), therefore immunohistochemistry was performed to help confirm the diagnosis. The following immunohistochemistry was performed: AE1/AE3, CA 19.9, BCL-10, CK7, CK20, NKX 3.1, Trypsin, Chymotrypsin, PSA, INSM-1, Chromo-A, Synaptophysin, and Ki-67.
The immunohistochemistry resulting positive included AE1/AE3, CA19.9, and BCL-10 (Figure 3). The Ki-67 was identified to have an increased proliferation rate (>70%), and the remaining immunohistochemical and special stains were negative. Therefore, the morphology and immunohistochemical profile were both confirmatory of an Acinic Cell Carcinoma.

Figure 1 (Low power magnification of cell block material)

Figure 2 (Higher magnification showing a cluster of pancreatic acinar cells with round nuclei and abundant finely granular cytoplasm.)

Figure 3 (BL10 immunohistochemistry stain, staining positive in the acinar cell population.)
Pancreatic acinar cell carcinoma (ACC) is a rare, aggressive exocrine tumor that is relatively still a mystery in the scientific community. Recent studies have identified frequent aberrant DNA methylation, abundant chromosomal amplifications and deletions suggesting defective DNA repair. Pancreatic ductal adenocarcinoma is the most common malignancy seen in the pancreas, however it is molecularly distinct from Acinar Cell Carcinoma as Pancreatic ductal adenocarcinoma has no recurrent point mutations. Studies have shown the tumor suppressor genes ID3, ARID1A, APC and CDKN2A as potential drug targets for Acinic Cell Carcinoma of the pancreas. Currently the gold standard of treatment is surgical resection, if permitting, along with gemcitabine based chemotherapy or radiofrequency ablation. In this case, the elected approach was chemotherapy, and in April 2024, CT abdomen is measuring the mass to be 5.4 cm which significantly decreased in size compared to October 2023 imaging. Currently the patient is doing well, and the plan is for total pancreatectomy with splenectomy and vascular reconstruction 3 weeks after the last day of chemotherapy.
The patient is a 47 year old male with no significant past medical history who presents with abdominal pain, chest pain, and dyspnea. Imaging showed a 24cm abdominal mass with encasement of the aorta, infrarenal IVC and right ureter invasion, bladder thickening, pulmonary embolisms, lymphadenopathy, and heterogenous attenuation of sacrum and pelvic bones. Prior to admission, the patient stated he had worsening abdominal distension, low back pain, weight loss, fevers, night sweats, and bloody urine for the past couple of months. Fine needle aspiration (FNA) and core needle biopsy of the abdominal mass was insufficient for definitive diagnosis and core needle biopsy of left supraclavicular lymph node was performed.
Cytologic description
Touch preparations of the core needle biopsy of the left supraclavicular lymph node showed discohesive cells with enlarged, irregular nuclei, fine chromatin, prominent nucleoli, and scant to moderate cytoplasm. The background showed lymphocytes and a tigroid background. The H&E of the core needle biopsy showed similar findings; however, the tigroid background is no longer appreciated. An extensive immunhistochemical (IHC) workup was performed and showed the tumor cells were weakly positive for pancytokeratin and CK7, but CK20 negative. PAX8, GATA3, TTF1, synaptophysin, chromogranin A, SOX10, Melan-A, myogenin, MyoD1, CD30, and Glypican-3 were negative. CD45 highlighted background lymphocytes. SALL4, PLAP, OCT3/4 were positive. The cytomorphology, histomorphology, and immunophenotype together are consistent with a germ cell tumor and a diagnosis of seminoma was rendered. Further workup revealed an ill-defined, heterogeneous area in the superior aspect of the right testis and an adjacent 0.7cm isoechoic focus with a hypoechoic rim. Chemotherapy regimen was initiated as the patient was not considered a surgical candidate.



Seminoma is a malignant germ cell tumor that derives from cells similar to primordial germ cells/gonocytes found in early embryonic development. Seminomas arise from the testis and are immunophenotypically and morpholocigally identical to germinomas and dysgerminomas that arise from the ovary, mediastinum, and pineal gland. Seminoma is the most common germ cell tumor of the testis and accounts for >50% of testicular germ cell tumors. Seminoma has a predilection for young men (37-41 years old); however, it can also be seen in men > 50 years old. Patients often present with a self-identified mass and vague discomfort in the testis. Less than 3% present with symptoms related to retroperitoneal metastasis such as lower back pain. Metastasis occurs via lymphatics to retroperitoneal lymph nodes, then to mediastinal and cervical lymph nodes.
Cytologically, seminomas appear as discohesive cell groups, flat sheets and individually with round to oval nuclei, fine chromatin, and prominent nucleoli. Cells have moderate cytoplasm that is delicate with vacuoles containing glycogen. The background often has a lymphocytic infiltrate and the characteristic tigroid pattern resulting from fragmented cytoplasm containing glycogen. Seminomas stain positive for SALL4, OCT3/4, PLAP, but are negative for AE1/AE3, CD30, AFP, and EMA.
References
1. BlueBooksOnline. tumourclassification.iarc.who.int. Accessed April 5, 2024. https://tumourclassification.iarc.who.int/chaptercontent/36/119
2. Cibas ES, Ducatman BS. Cytology: Diagnostic Principles and Clinical Correlates. Fifth edition. Elsevier; 2019.
Richard Judelson, MD, Wafaa Elatre, MD, MPH
Clinical History
59 year-old female who was recently diagnosed with solid papillary carcinoma of the left breast and is status post lumpectomy, adjuvant radiation and hormone therapy. Surveillance MRI identified a 2.9 cm left supraclavicular mass just below the thyroid gland, which was suspicious for lymphadenopathy vs a thyroid or parathyroid nodule. Follow up ultrasound of the head & neck confirmed an ovoid shaped, solid mass inferior to the thyroid gland which appears to be contiguous with the left thyroid and devoid of calcifications. This mass measures 2.4 cm and is consistent with TIRADS 4. The patient subsequently underwent ultrasound-guided fine needle aspiration of the neck mass.
Cytomorphologic findings
Cellular Diff-Quik and Papanicolaou stained smears showed abundant mature squamous cells with associated bacterial organisms. Rare bland follicular cells admixed with watery and thick colloid and mixed inflammatory cells were present in the background. The cell block showed mature squamous cells, bacterial organisms and mixed inflammation.
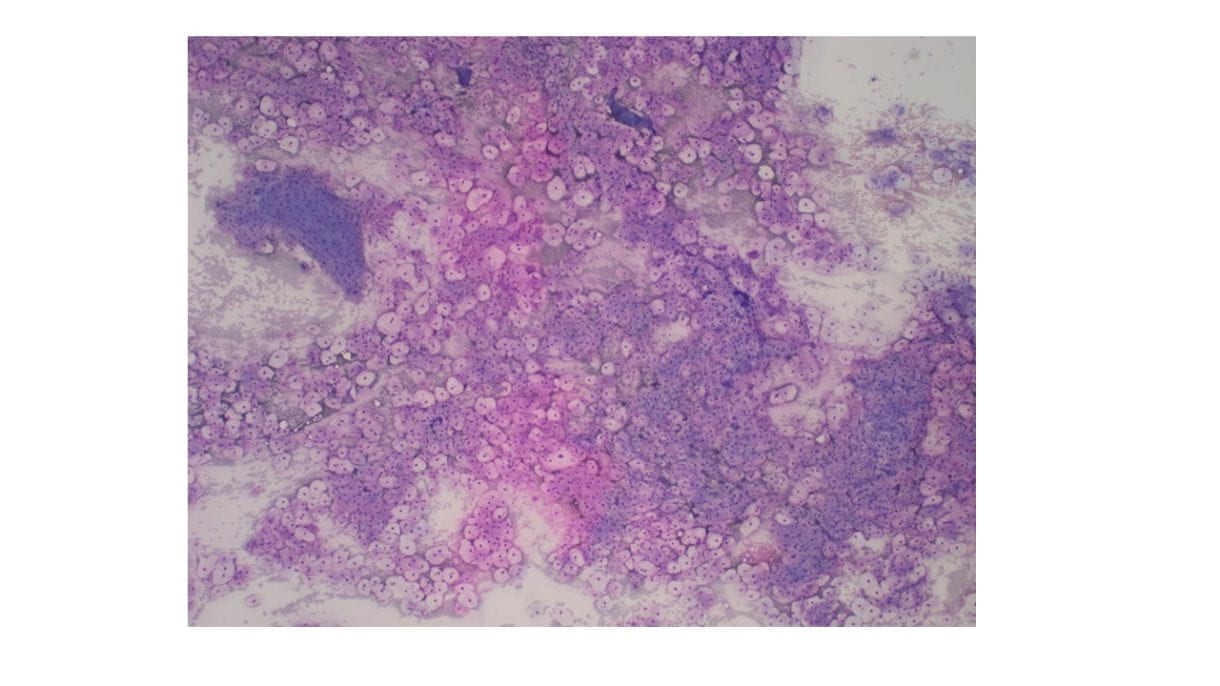
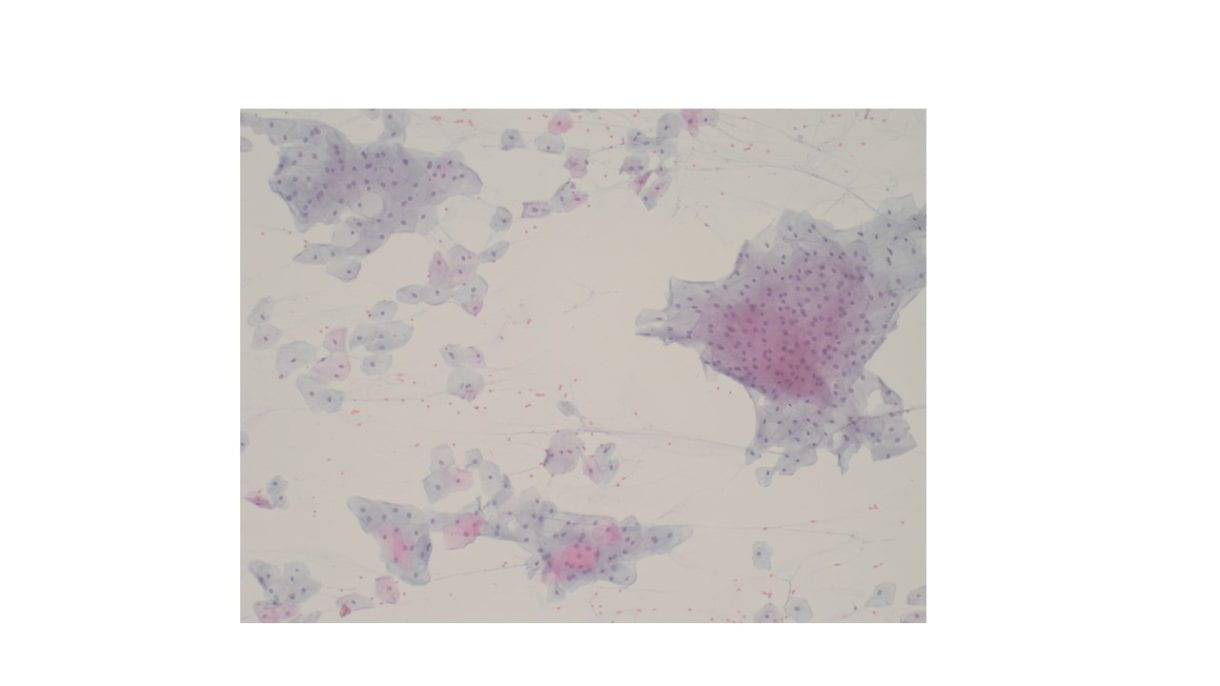
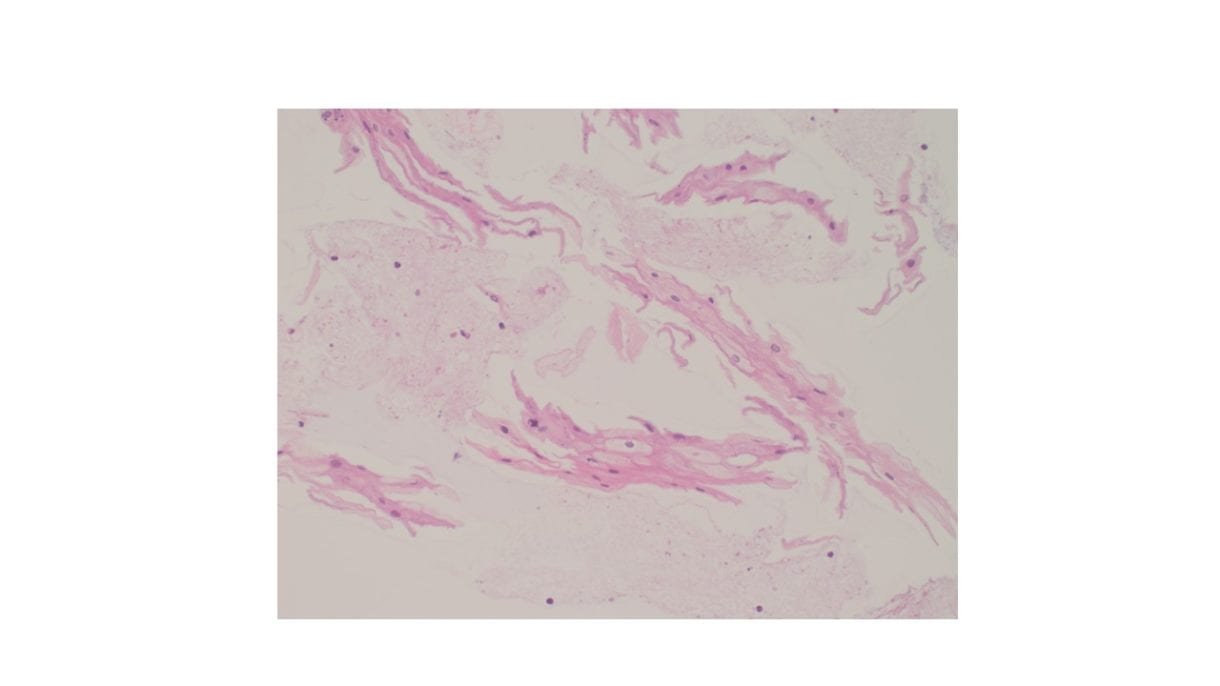

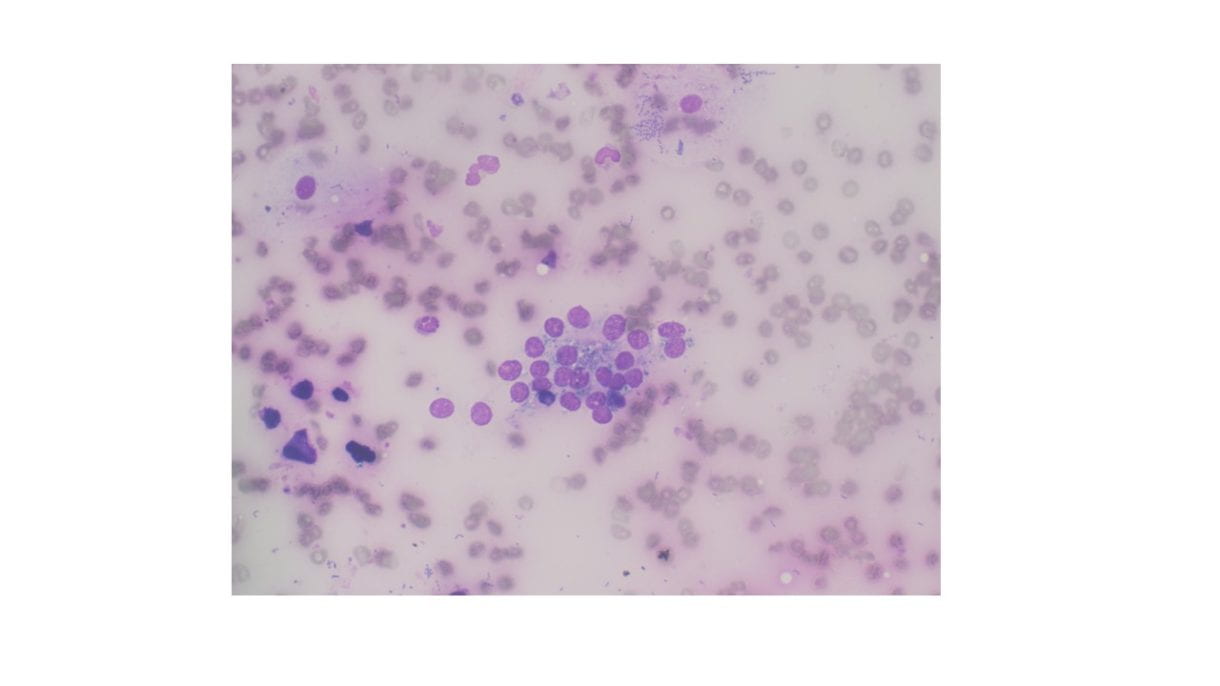
Discussion
Suspected thyroid nodules consisting almost entirely of mature squamous epithelial cells is rare. The differential diagnosis of benign appearing squamous epithelium in the thyroid includes squamous metaplasia, thyroglossal duct cyst, thymic/lymphoepithelial/ultimobranchial body remnants, epidermoid/dermoid cysts or esophageal tissue (accidental esophageal sampling vs esophageal diverticula). Any squamous atypia should raise suspicion of a malignant entity such as papillary thyroid carcinoma with squamous changes or a metastatic squamous cell carcinoma.
Differentiating between the various benign squamous lined cystic lesions in and around the thyroid is challenging and definitive diagnosis is best reserved for excisional biopsy.
Of note, the presence of mostly bland, mature squamous epithelium associated bacteria and mixed inflammation is highly suspicious for sampling of esophageal mucosa. In such a case, recommending radiologic follow up with barium studies may be helpful to rule out the possibility of an esophageal diverticula.
Reference
Chen AL, Renshaw AA, Faquin WC, Alexander EK, Heller HT, Cibas ES. Thyroid FNA biopsies comprised of abundant, mature squamous cells can be reported as benign: A cytologic study of 18 patients with clinical correlation. Cancer Cytopathol. 2018 May;126(5):336-341. doi: 10.1002/cncy.21976. Epub 2018 Apr 10. PMID: 29634853.
CLINICAL HISTORY:
The patient is 65-year-old male with no past medical history who presented with anorexia and intermittent back and abdominal pain for few weeks. No other symptoms, such as nausea or vomiting, were reported. Physical examination did not reveal any specific findings. There was no family history of gastrointestinal diseases. The computed tomography (CT) scans showed 3.4 cm infiltrative pancreatic uncinate mass. No other masses or metastasis were observed on the scan. The patient was scheduled for endoscopy to perform endoscopic ultrasound fine-needle aspiration (EUS-FNA) to the mass.
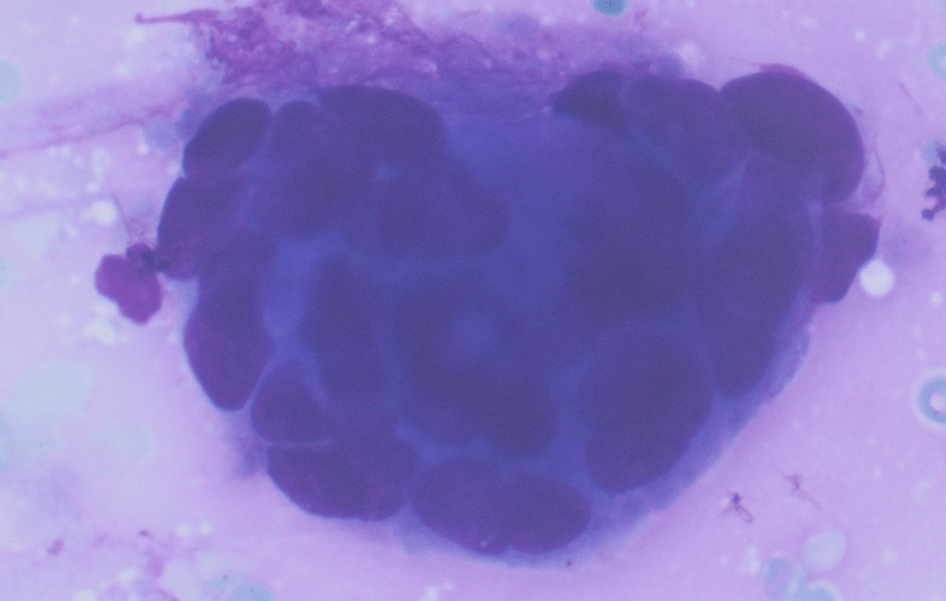
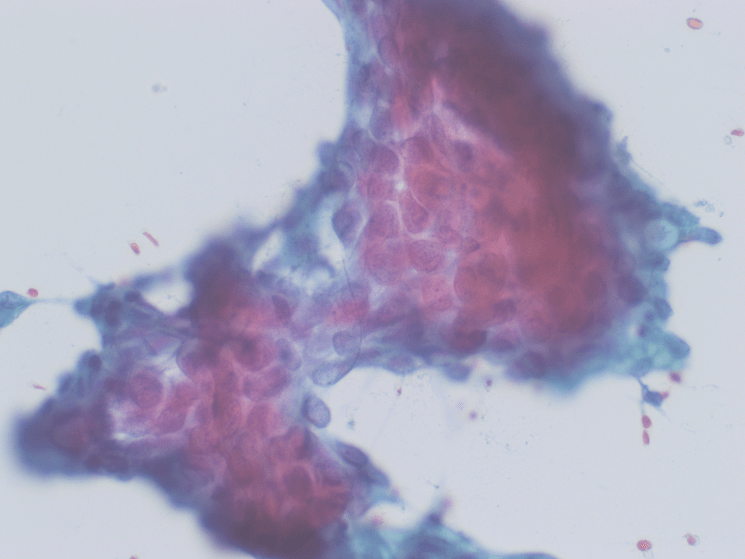
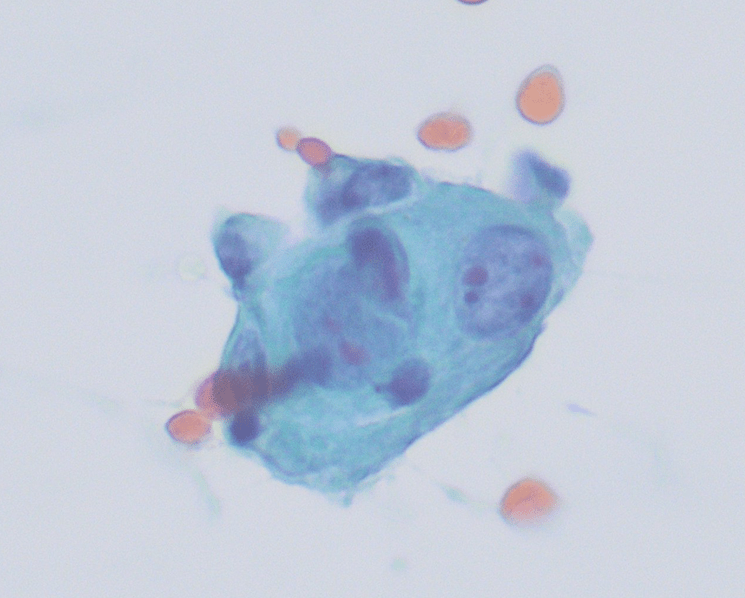
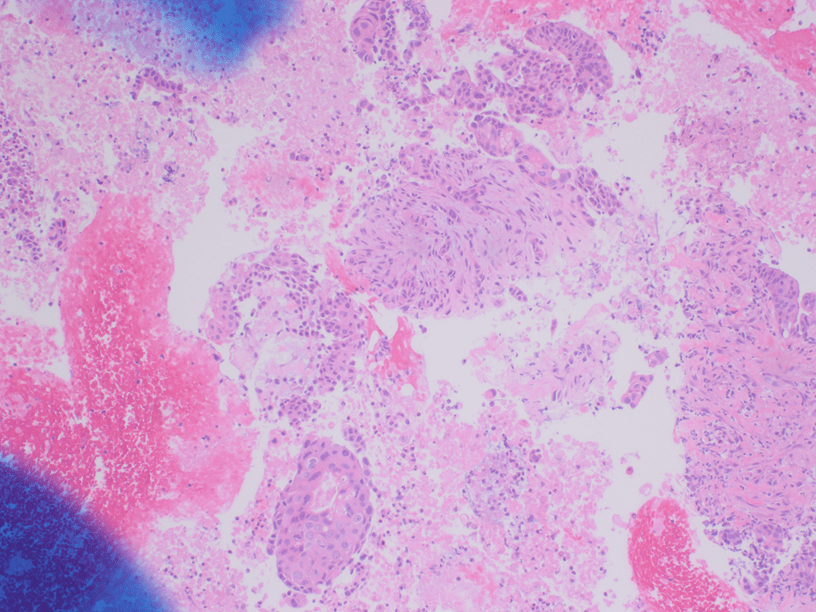
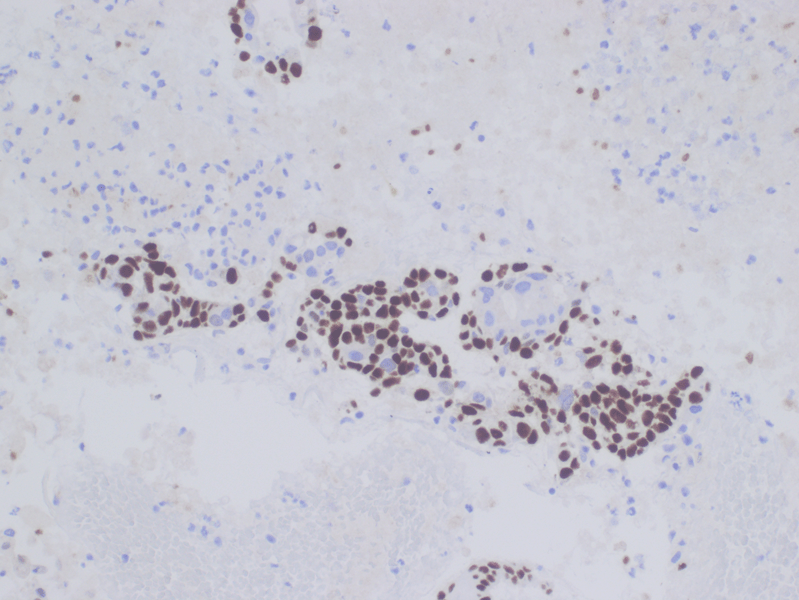
The Diff-Quik and Pap-stained smear preparations were cellular, showing Irregularly sized and shaped cohesive cluster and singly discohesive, dispersed cells. The clusters showed uneven distribution of cells. The cells showed irregular nuclear contours, nuclear enlargement, hypochromasia and hyperchromasia, anisonucleosis (greater than 4:1 variation in diameter within a single cluster), irregular chromatin distribution, and focal vacuolated cytoplasm. Few clusters showed focal dense blue cytoplasm in Diff-Quik and blue to red dense cytplasim in Pap stain. The H&E sections from cell block showed clusters with dense pink cytoplasm intimately associated with disorganized angulated glands and single cells in desmoplastic and myxoid stroma. The cells in H&E show similar cytomorphologic findings to smear preparations.
The initial differential diagnosis was pancreatic ductal adenocarcinoma (PDAC) & its variants/subtypes, reactive/reparative atypia, chronic pancreatitis, radiation changes, and metastatic carcinoma.
Immunohistochemical stains were performed and showed positive for CK7, p63 (focal, see fig 5), and CK20 (rare cells). The cells were negative for TTF-1, SATB2, and CDX2.
Diagnosis:
The histomorphology and immunohistochemical findings fit Adenocarcinoma with focal area of squamous differentiation consistent with adenosquamous carcinoma of the pancreas.
The patient underwent pancreaticoduodenectomy (Whipple procedure). The resection showed similar histology to cell block H&E and was ultimately diagnosed as Adenosquamous carcinoma of the pancreas.
Discussion:
Pancreatic adenosquamous carcinoma is a rare aggressive subtype of ductal adenocarcinoma and accounts for 1-4% of all exocrine malignancies of the pancreas. Also referred to as mucoepidermoid carcinoma and adenoacanthoma in older literature, adenosquamous carcinoma demonstrates both malignant squamous cell and glandular differentiation. In surgical pathology and per WHO 5ed, the squamous component arbitrarily should account for ≥ 30% of the neoplasm in order for it to qualify as adenosquamous. Squamous metaplasia of the pancreatic ductal epithelium occurs most commonly in the setting of chronic pancreatitis but is noted in the adjacent ducts of only about 4% of adenocarcinomas. Squamous metaplasia of pre-existing adenocarcinoma has been suggested by some authors as a mechanism underlying the histogenesis of pancreatic adenosquamous carcinoma. Making a diagnosis of adenosquamous carcinoma on fine needle aspiration before surgery can be difficult and it is possible when aspirates show evidence of both squamous and glandular differentiation. Adenosquamous carcinoma also may represent a metastasis to the pancreas arising from lung, colon, esophagus, salivary glands, and female reproductive organs. Pure squamous cell carcinomas of pancreas are exceedingly rare and findings of malignant squamous cells in FNA should prompt the pathologist to search for glandular differentiation for adenosquamous carcinoma. The adenosquamous carcinoma could be recognized/suggested in cytology by unique cytological features that recapitulate the histology. The squamous component often expresses p63, p40, and low-molecular-weight cytokeratins. Almost all cases harbour KRAS mutations at codon 12, and they show highly enriched TP53 mutations, along with 3p loss and mutations in the RNA surveillance gene UPF1. Immunohistochemically, they show loss of p16 (CDKN2A) protein expression, loss of SMAD4 (DPC4) protein, and strong nuclear p53 immunoreactivity, which is similar to the molecular signature found in Pancreatic ductal adenocarcinomas (PDACs). Adenosquamous carcinomas appear to have a worse prognosis than pure ductal adenocarcinomas even when resected, with a median survival time of about 9 months. The presence of any squamous component in the neoplasm appears to portend a worse prognosis.
REFERENCES:
Luna Li, MD, PhD, Wafaa A. Elatre, MD, MPH
– 59 year old male with medical history of diabetes mellitus, hypertension, heart failure with reduced ejection fraction, and possible malignancy.
Patient found to have pleural effusion as well as possible mass in bronchus and diffuse lymphadenopathy in mediastinum.
Had diagnostic thoracentesis on 10/28/22. Diagnosis “positive for carcinoma” with unknown origin on cytology
Immunoreactivity for GATA3, CK7 and mammaglobin are in favor of a breast primary, while a urothelial cell origin cannot be entirely ruled out.
Biopsy of right bronchus intermedius mass on 11/1/2022 was performed. Pathology diagnosis was well-differentiated squamous carcinoma with an atypical immuophenotype. The cells express GATA-3 which is a marker or urothelium.
• Patient developed pericardial effusion one month later. Patient also reports cough productive of white sputum, bilateral extremities swelling x1 week, and 20 lb weight loss over 1 month with decreased appetite. His shortness of breath is aggravated by ambulation. Patient has worked in construction for many years.
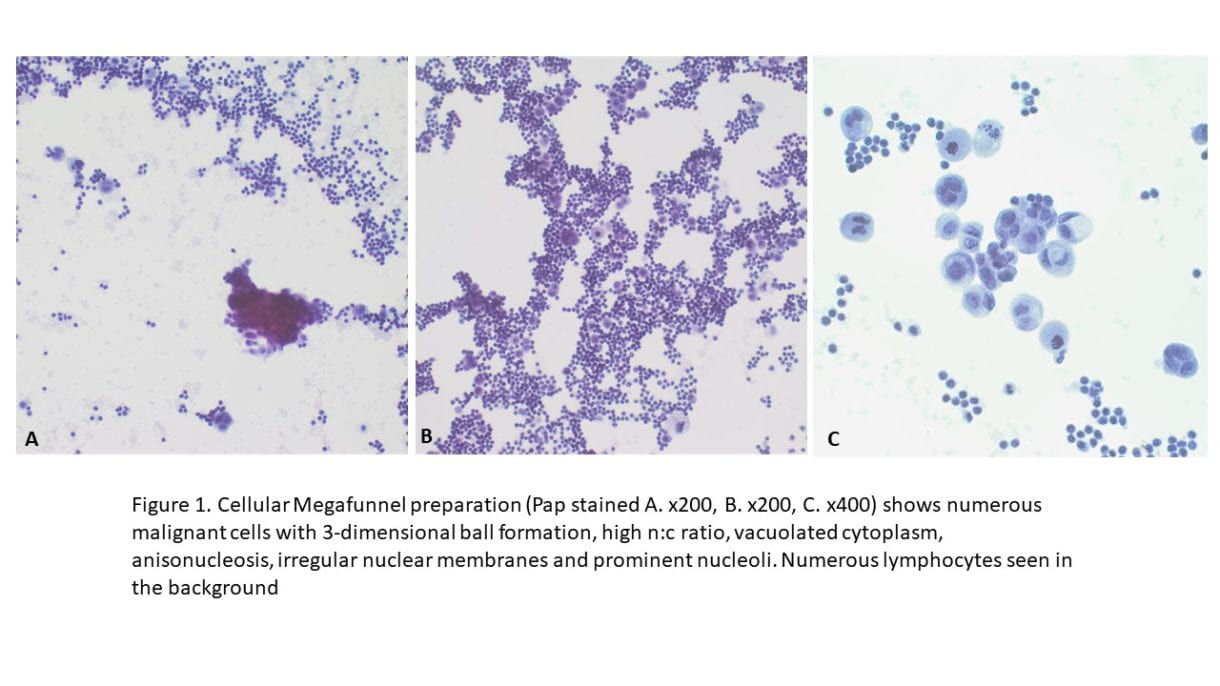
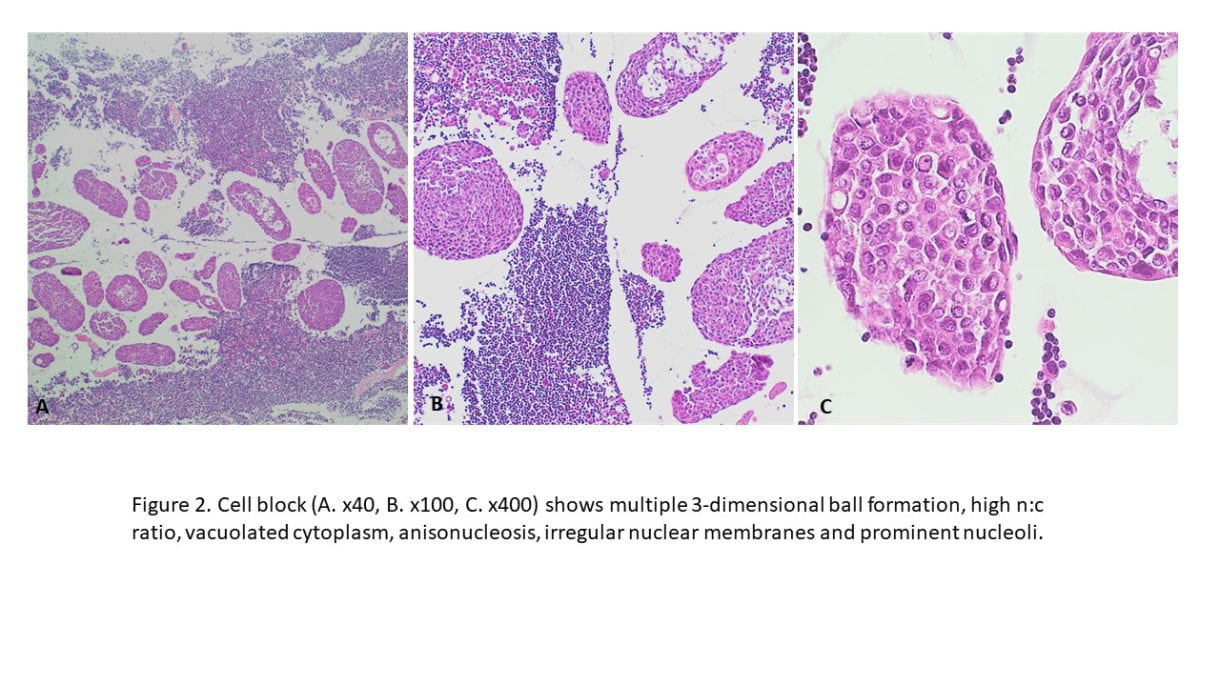
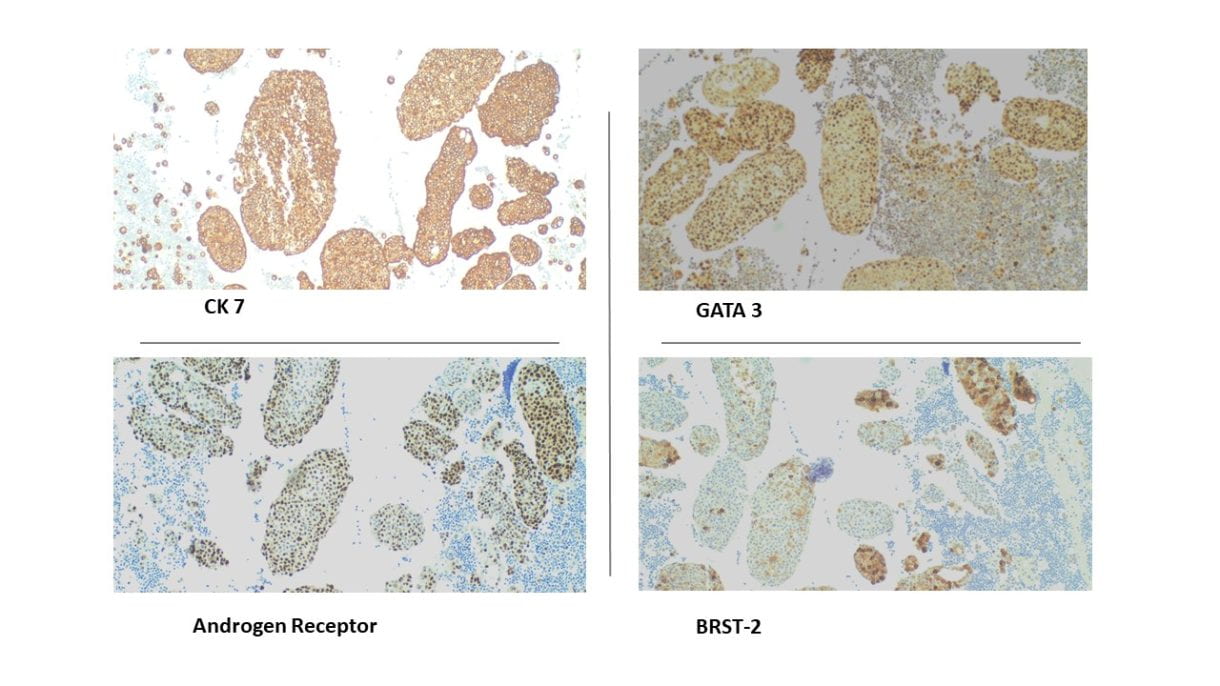
•Immunohistochemical staining were performed on the cell block to further characterize the lesion. CK7, GATA3, BRST-2, and Androgen Receptor were positive on tumor cells, CK5/6 and p16 were focally positive, while CK20, Mammaglobin, S100, SOX10, Uroplakin II, Calretinin, TTF-1 and Napsin A showed negative staining on tumor cells. Special staining for Mucicarmine was essentially negative.
Molecular testing
•Tempus Labs xT 648 gene panel performed on surgical biopsy specimen Right bronchus intermedius mass.
•xT 648 gene panel focused on actionable mutations by DNA sequencing.
•PIK3CA mutation p.I102_P104delinsKS with an inframe deletion – GOF (exon 1) was identified.
Discussion
•Primary pulmonary salivary gland–type cancer is rare and comprises less than 1% of all lung tumors.
•This group of tumors derives from small salivary glands in the respiratory system and mainly include two common histological subtypes of mucoepidermoid carcinoma and adenoid cystic carcinoma.
• Like salivary duct carcinoma, pulmonary salivary gland–type carcinoma is positive for androgen receptor (AR), GATA 3 is usually positive, and most cases are also positive for GCDFP-15 (BRST-2).
•HER2/neu expression is usually seen but diffuse strong membranous staining or HER2 amplification on FISH is seen in only 25% of cases.
A novel mutation in a pulmonary salivary-type duct carcinoma with a PIK3CA mutation was identified which could guide treatment options for this rare cancer
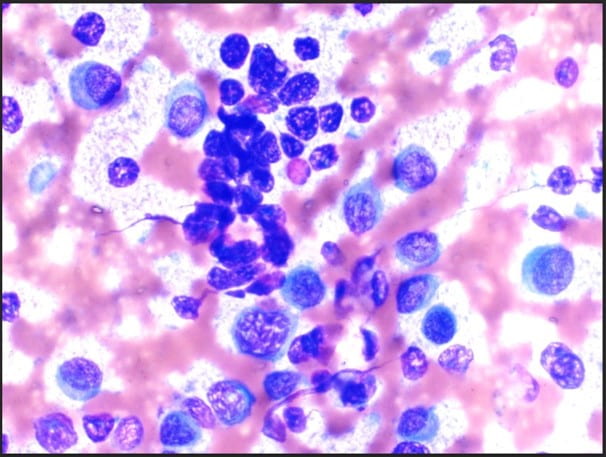 Figure 1. The cellular smear preparation demonstrates singly dispersed and clustered large immature plasma cells with eccentric nuclei, coarse chromatin, prominent eosinophilic nucleoli, high nuclear-to-cytoplasmic ratio, and scant to moderate amounts of cytoplasm. Admixed with smaller mature plasma cells and normal blood elements (Diff-Quik, 40X).
Figure 1. The cellular smear preparation demonstrates singly dispersed and clustered large immature plasma cells with eccentric nuclei, coarse chromatin, prominent eosinophilic nucleoli, high nuclear-to-cytoplasmic ratio, and scant to moderate amounts of cytoplasm. Admixed with smaller mature plasma cells and normal blood elements (Diff-Quik, 40X).
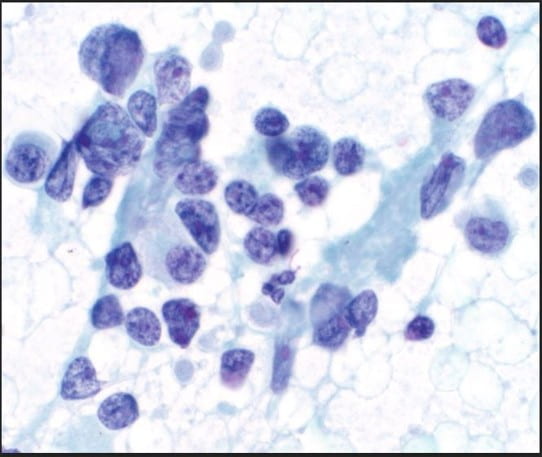 Figure 2. The cellular smear preparation demonstrates mostly clustered immature plasma cells, coarse chromatin, prominent eosinophilic nucleoli, high nuclear-to-cytoplasmic ratio, and scant to moderate amounts of cytoplasm. At the periphery of this grouping are smaller mature plasma cells. The Pap stain highlights the prominent eosinophilic nucleoli and the granularity of the coarse chromatin (Pap stain, 40X).
Figure 2. The cellular smear preparation demonstrates mostly clustered immature plasma cells, coarse chromatin, prominent eosinophilic nucleoli, high nuclear-to-cytoplasmic ratio, and scant to moderate amounts of cytoplasm. At the periphery of this grouping are smaller mature plasma cells. The Pap stain highlights the prominent eosinophilic nucleoli and the granularity of the coarse chromatin (Pap stain, 40X).
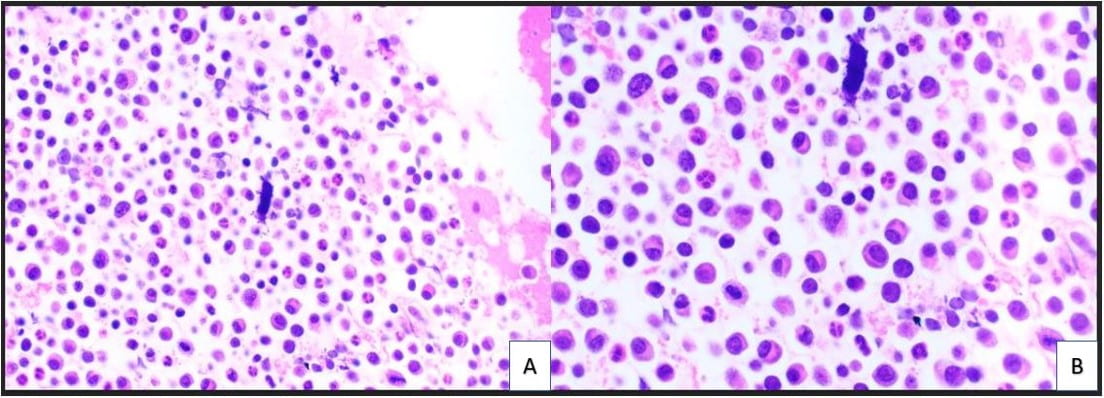 Figure 3. The cell block redemonstrates features seen in the smear preparations with more variably sized plasma cells and blood elements including neutrophils (A: H&E, 10X; B: H&E, 20X).
Figure 3. The cell block redemonstrates features seen in the smear preparations with more variably sized plasma cells and blood elements including neutrophils (A: H&E, 10X; B: H&E, 20X).
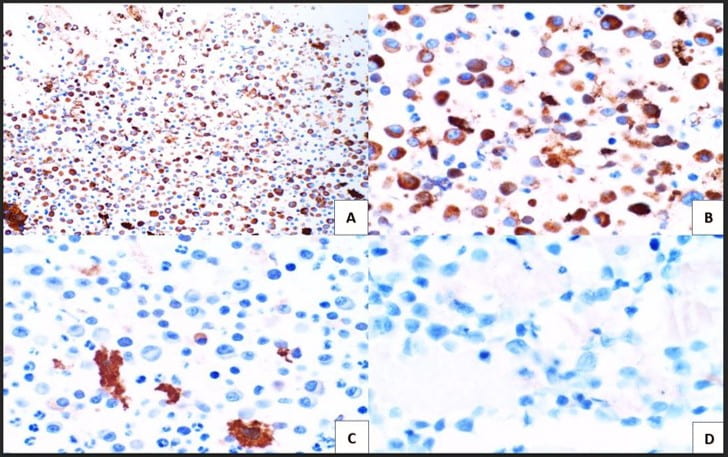 Figure 4. Immunohistochemical studies confirm plasma cell lineage with CD138 (A) strongly positive in both the larger immature and smaller immature plasma cells (10X). Kappa (B) highlights a majority of the plasma cells (20X). While lambda (C) is positive in only a few plasma cells. CD30 (D) is completely negative as is c-Myc (not pictured) (20X).
Figure 4. Immunohistochemical studies confirm plasma cell lineage with CD138 (A) strongly positive in both the larger immature and smaller immature plasma cells (10X). Kappa (B) highlights a majority of the plasma cells (20X). While lambda (C) is positive in only a few plasma cells. CD30 (D) is completely negative as is c-Myc (not pictured) (20X).
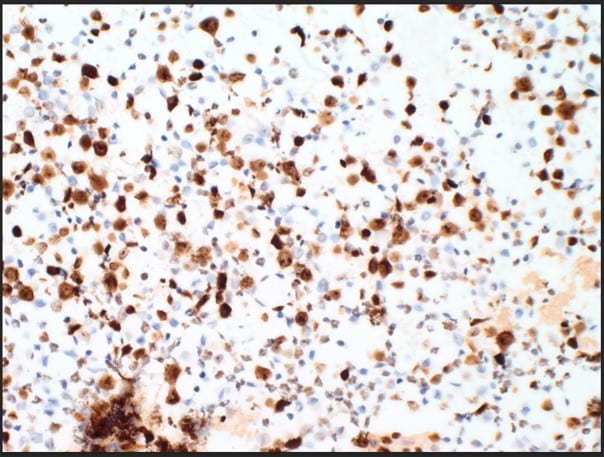 Figure 5. The proliferation rate via Ki-67(MIB-1) within the plasma cells is exceedingly high approximating >80-90% (10X).
Figure 5. The proliferation rate via Ki-67(MIB-1) within the plasma cells is exceedingly high approximating >80-90% (10X).
The cytologic features of mostly immature plasma cells (Figures 1-3) as confirmed by ubiquitous CD138 positivity points towards a plasma cell dyscrasia (Figure 4). A high number of the plasma cells show positivity by kappa immunostaining as compared to a significantly lower number of few plasma cells showing positivity by lambda immunostaining; thereby proving kappa restriction (Figure 4). Not pictured in our treatise, but associated with plasma cell dyscrasias, are Russell bodies (hyaline cytoplasmic inclusions containing immunoglobulin) and Dutcher bodies (intranuclear cytoplasmic invaginations). Russell bodies are seen in both benign and malignant plasma cells. Yet, Dutcher bodies are more common in neoplastic plasma cells. CD30 was negative, but expression can be used to decide anti-CD30 targetability in multiple myeloma. c-Myc, which is negative, is assessed as overexpression is associated with adverse clinical outcomes. Ki-67(MIB-1) is high (Figure 5), which is indicative of the aggressive proliferation of the plasma cell neoplasm. Table 1 summarizes interpretation of the immunostains.
The bone marrow biopsy was used for further molecular and cytogenetics testing which allowed for stratification of risk, prognosis, and treatment.
Molecular studies/cytogenetic testing from subsequent bone marrow biopsy of the same site showed the following:
Positive for IGH/FGF3 rearrangement [FISH].
Positive for IGH/CCND1 rearrangement [FISH].
Positive for 1q gain, gain of 9 and 13q deletion/monosomy 13 [FISH].
Negative for TP53 (17p13) deletion, and for rearrangements in IGH/MAF or IGH/MAFB [FISH].
Multiple myeloma has a poor prognosis especially with TP53(17p13) deletion (a high risk chromosomal abnormality). From diagnosis, most patients live from less than 6 months to 10 years with the median survival of 5.5 years. Prognosis is determined by the combination of two risk stratification components: genetics and revised multiple myeloma staging system. The Revised International Staging System (R-ISS) for Multiple Myeloma was devised by the International Myeloma Working group incorporating these two risk stratification components to categorize multiple myeloma patients into three (3) major stages. These were later recommended by NCCN and are standard of practice.
Stage I criteria include (1) beta-2 microglobulin ≤3.5 g/dL and albumin ≥3.5 g/dL, (2) standard risk for chromosomal abnormalities (CA), and (3) normal lactate dehydrogenase (LDH) levels. Stage II is an intermediate category for patients whose clinical and cytogenetic profile are do not meet criteria for either Stage I or Stage III. Stage III criteria include (1) having a beta-2 microglobulin ≥ 5.5 g/dL and (2) being high risk for CA or having a high LDH.
From diagnosis, most patients live from less than 6 months to 10 years with the median survival of 5.5 years. The R-ISS further stratifies this by associating each stage with a specific median progression-free survival (PFS). Patients in Stage I have a median PFS of 66 months. Median PFS for Stage II and Stage III are 42 months and 29 months, respectively. The patient is Stage II based on his beta-2 microglobulin levels and standard/intermediate risk for chromosomal abnormalities.
Cibas ES, Ducatman BS. Cytology: Diagnostic Principles and Clinical Correlates. Philadelphia, PA: Elsevier; 2021.
DeMay RM. The Book of Cells: A Breviary of Cytopathology. American Society of Clinical Pathology: 2016.
Callander NS, Baljevic M, et al. NCCN Guidelines® Insights: Multiple Myeloma, Version 3.2022. J Natl Compr Canc Netw. 2022;20(1):8-19.
Palumbo A, Avet-Loiseau H, et al. Revised International Staging System for Multiple Myeloma: A Report From International Myeloma Working Group. J Clin Oncol. 2015;33(26):2863-9.
Swerdlow SH, Campo E, Seto M, et al. (eds). WHO classification of Tumours of Haematopoietic and Lymphoid Tissues. 4th ed. International Agency for Research on Cancer: 2017.

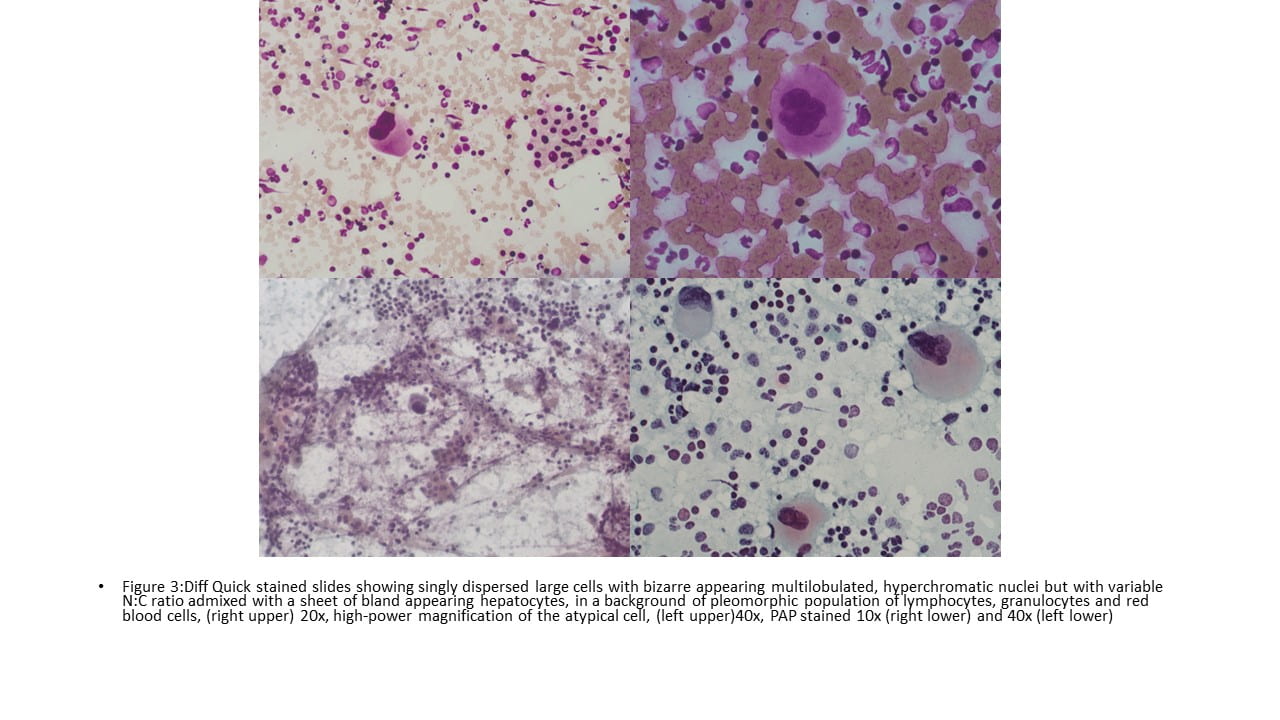
 IHC Stains
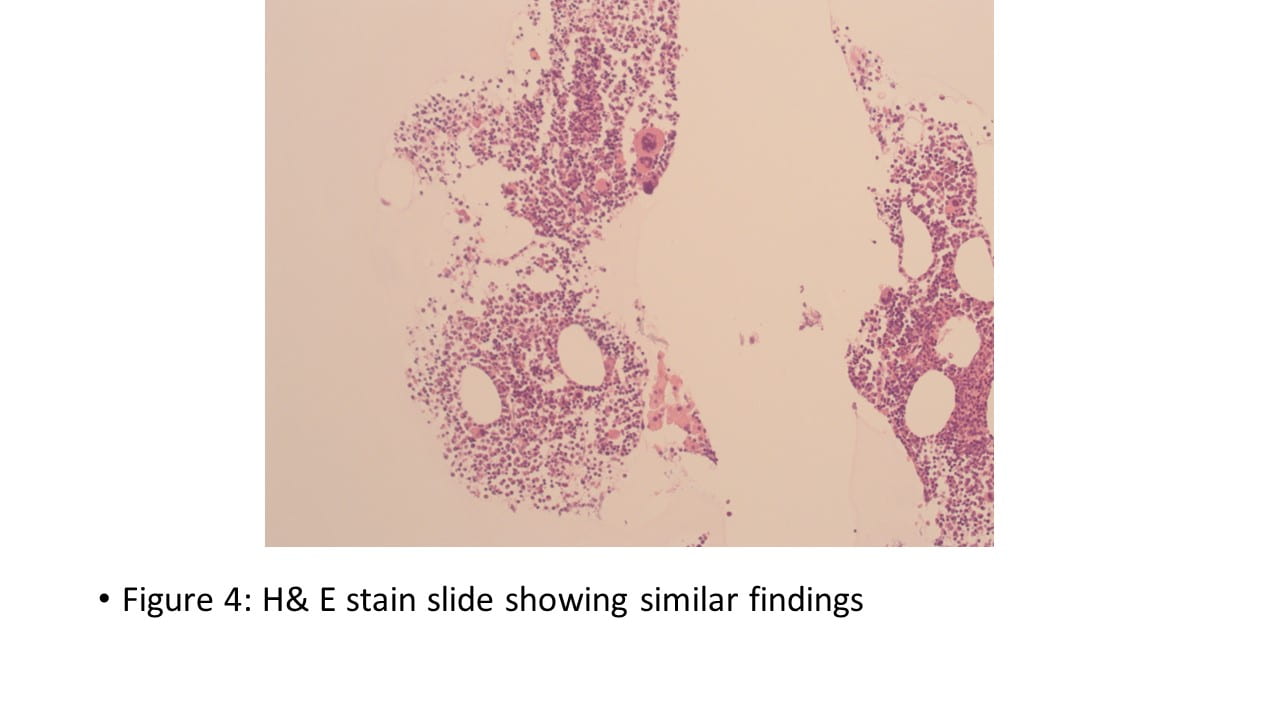
IHC Stains 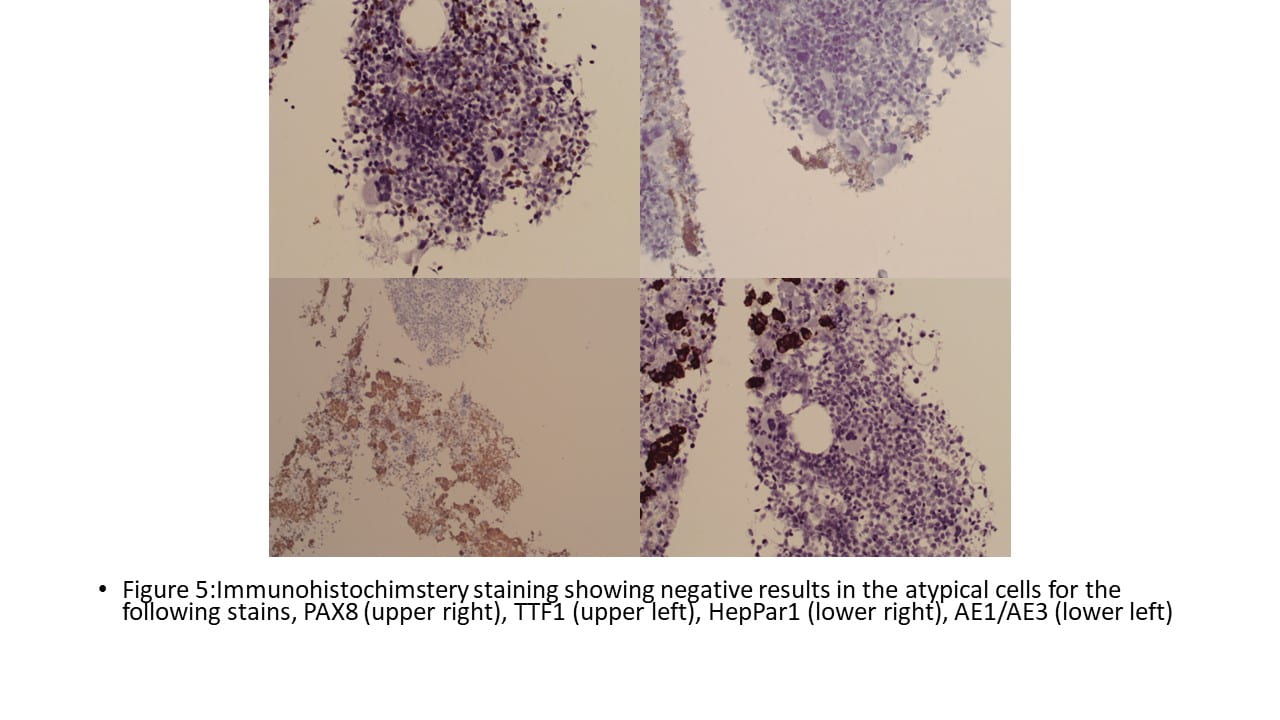
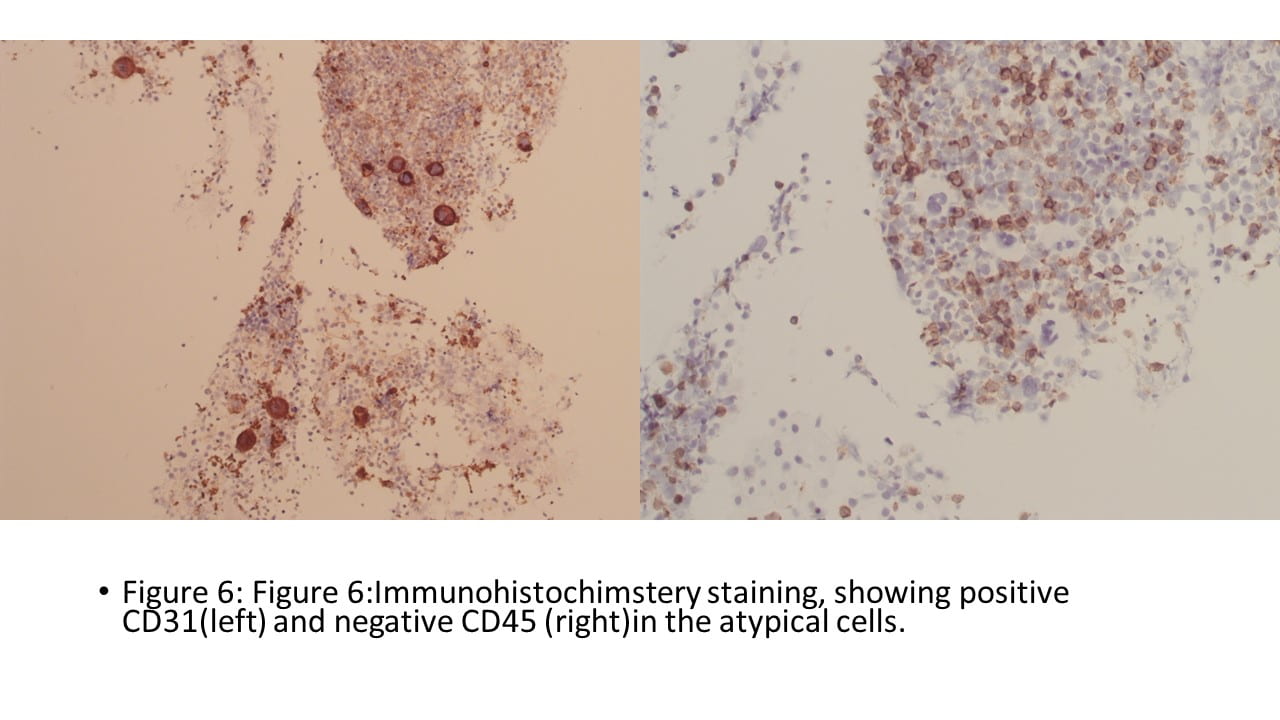 Final Cytologic diagnosis
Final Cytologic diagnosis1.Kapatia G, Kaur A, Rastogi P, et al. Extramedullary hematopoiesis: Clinical and cytological features. Diagnostic Cytopathology. 2020;48:191–196. https:// doi.org/10.1002/dc.24353
2.Bao Y, Liu Z, Guo M, Li B, Sun X, Wang L. Extramedullary hematopoiesis secondary to malignant solid tumors: a case report and literature review. Cancer Manag Res. 2018;10:1461-1470
https://doi.org/10.2147/CMAR.S161746
![]()
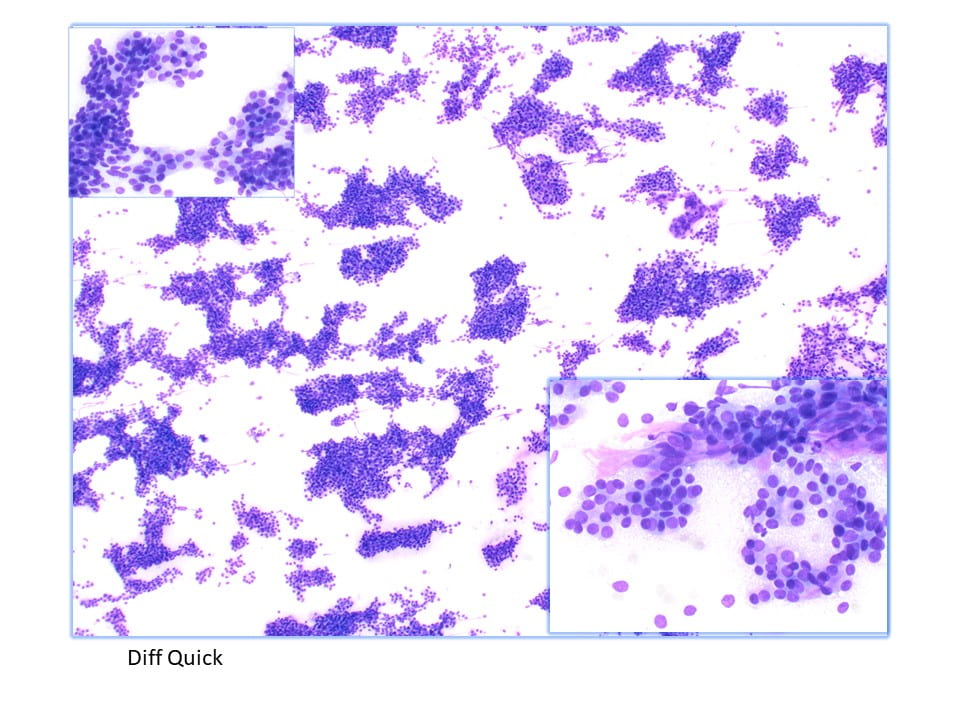
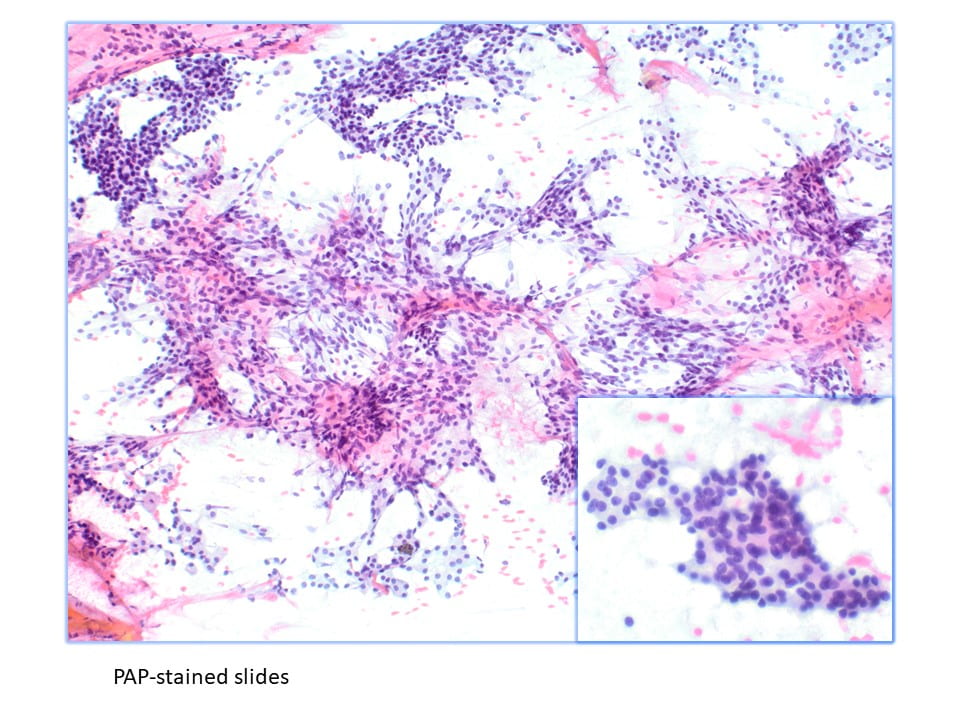
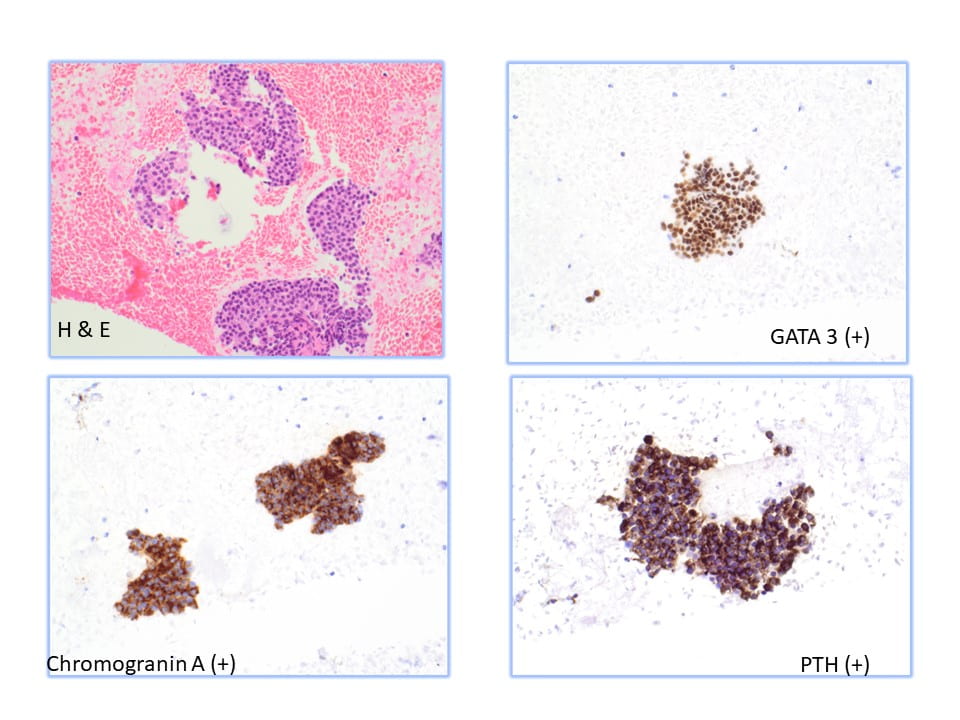
Parathyroid tissue on FNA can be difficult to identify due to overlapping features with thyroid neoplasms, particularly follicular neoplasms. Parathyroid tissue has been misdiagnosed as follicular thyroid carcinoma, papillary thyroid carcinoma, and medullary thyroid carcinoma on FNA. The sensitivity of FNA in the diagnosis of parathyroid tissue has been reported to be between 40% and 86%. The previous literature regarding FNA of the parathyroid gland has only characterized the cytomorphology on FNA smears. Characteristics such as high cellularity, the presence of naked nuclei, loose 2-dimensional clusters, and papillary architecture on FNA smears are suggestive of parathyroid tissue.