
Clinical History
![]()
The patient is a 34 year old male who presented with to the ED with a left calf mass. The patient first noticed the mass 3 years ago and it had been slowly increasing in size. Recently it had become more painful.
Physical exam demonstrated a large nontender calf mass with no overlying skin changes, induration or fluctuance. The patient was referred by MRI evaluation.
Initial Work Up
MRI of the lower extremity showed a 5.2 x 6.6 x 19.3 cm mass centered in the lateral (peroneal) muscular compartment of the left calf. The lesion abutted, but did not encircle, the anterior neurovascular bundle. The mass abutted the fibula but did not involve the osseous structures. Biopsy of the lesion was recommended for further evaluation.
The patient was sent for ultrasound guided biopsy with cytopathology at bedside for rapid on-site evaluation.
CYTOLOGY (Touch preparations of 14G core needle biopsy of the left calf mass)
The Diff-Quik stained touch preparations showed large clusters and numerous singly dispersed cells with large pleomorphic hyperchromatic nuclei, irregular nuclear contours, densely granular chromatin, prominent irregular macronucleoli and moderate to abundant amounts of vacuolated granular cytoplasm.
Many of the nuclei have no cytoplasm and are naked. The background is predominantly blood elements.
Figure 1. Touch Preparation with clusters of malignant cells with pleomorphic nuclei and abundant fragile, granular and vacuolated cytoplasm (Diff-Quik stain, 20X).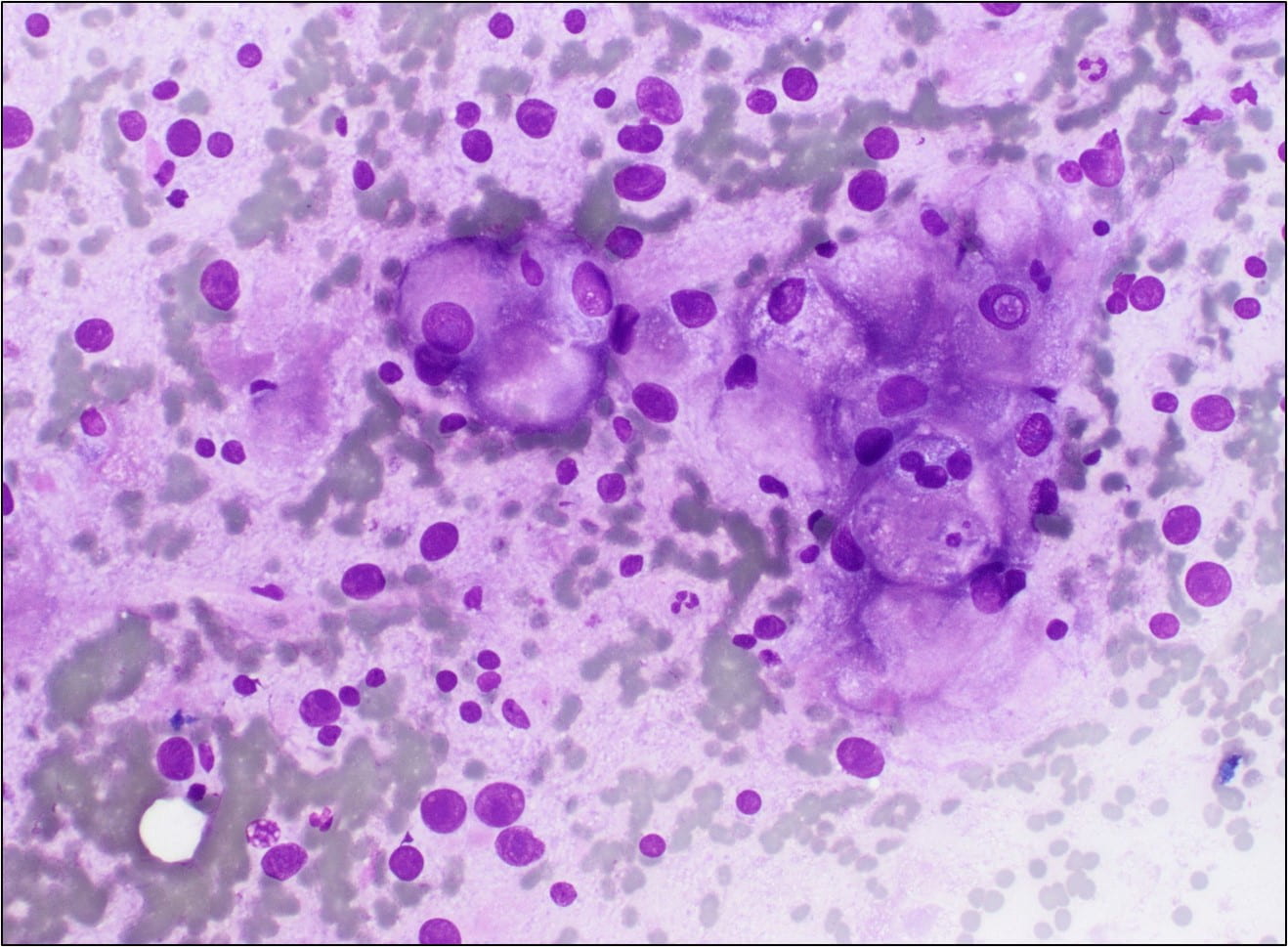
Figure 2. Many of the surrounding nuclei have irregular macronucleoli and are naked as the cytoplasmic contents have spilled into the background (Diff-Quik stain, 20X).
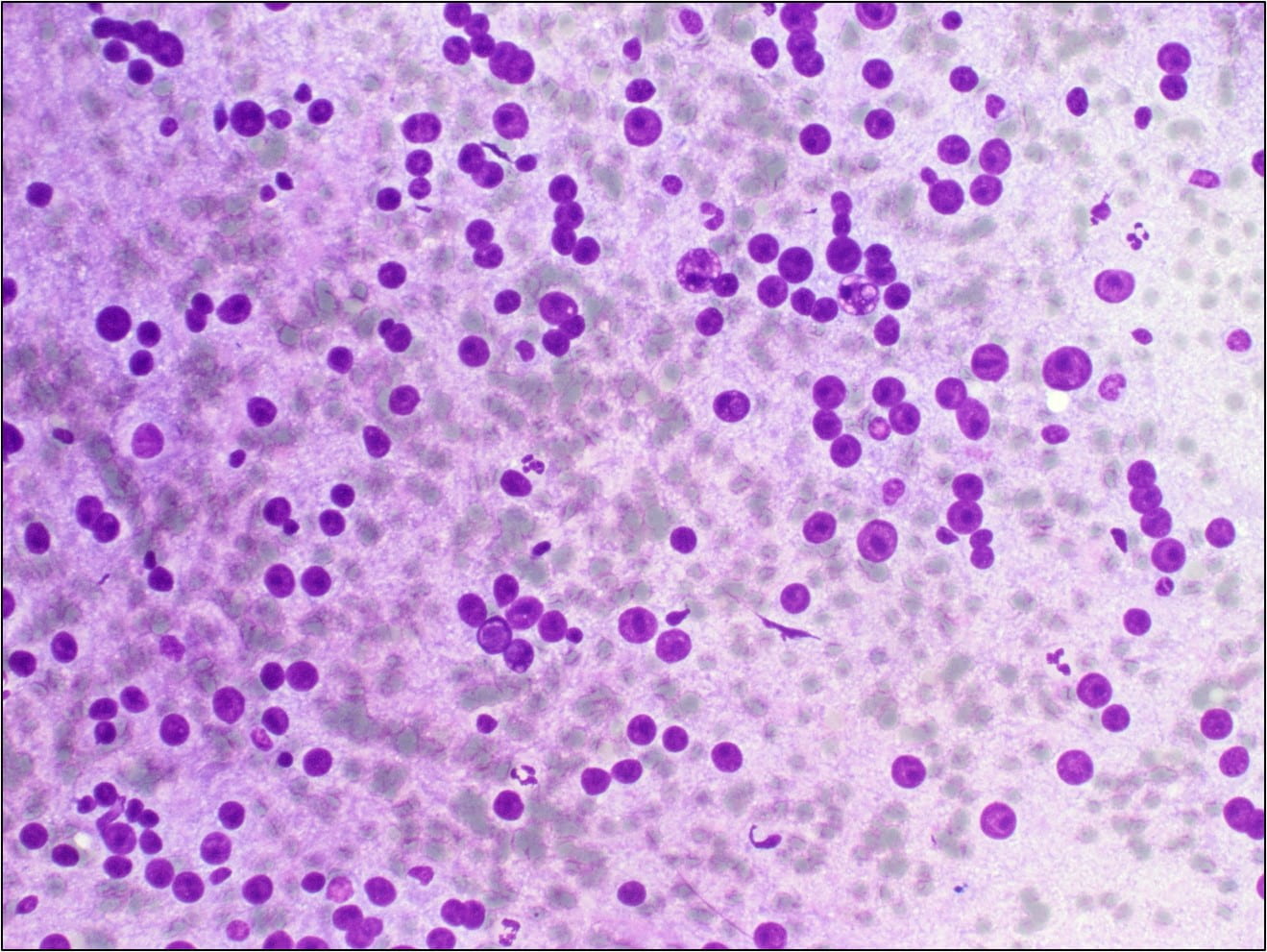
Figure 3. Some of the naked nuclei are markedly pleomorphic and surrounded by abundant granular disrupted cytoplasmic contents (Diff-Quik stain, 40X).
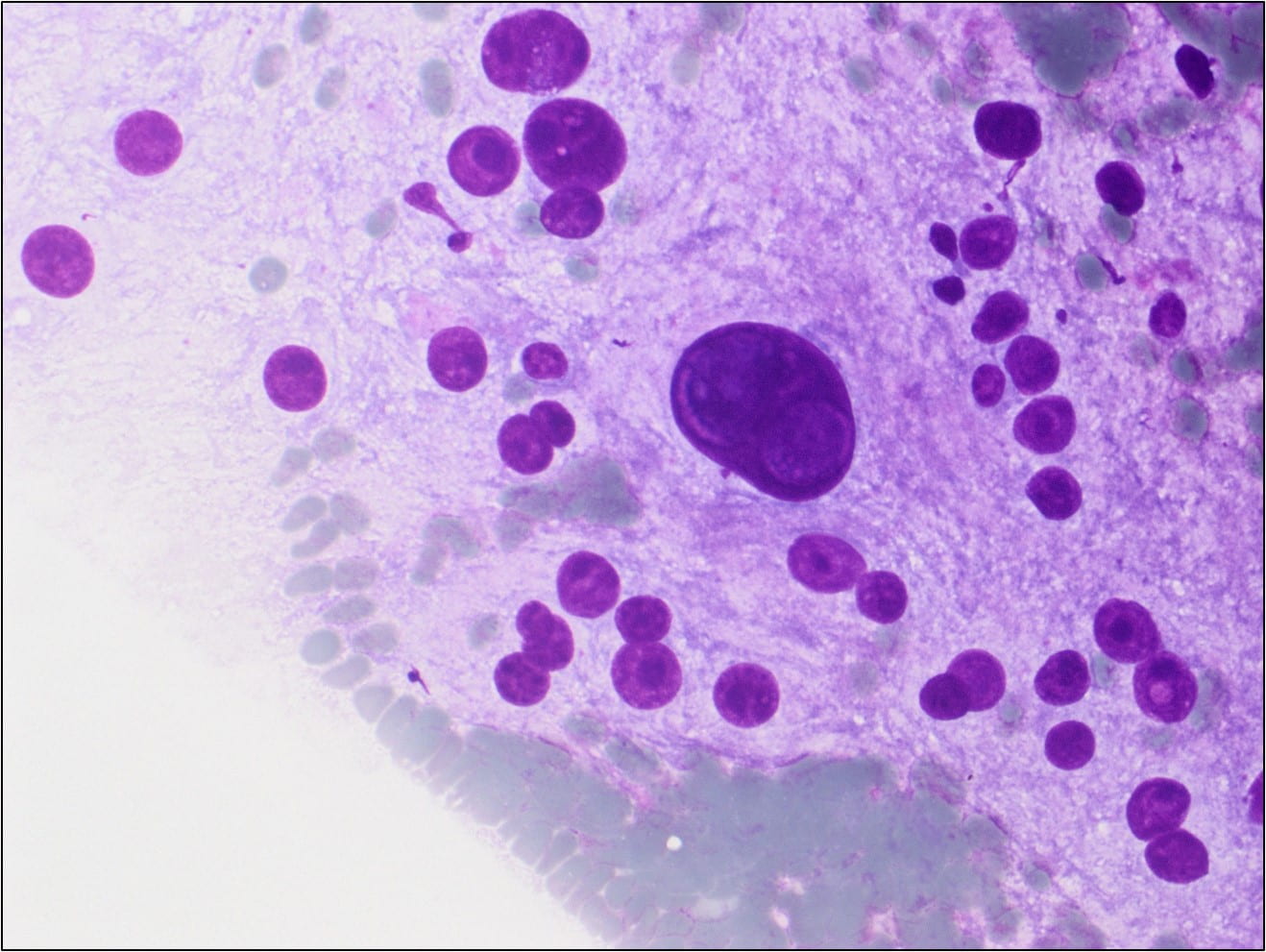
Figure 4. The tumor cells are arranged in aggregates which form dense trabeculae (Hematoxylin and eosin, 20X).
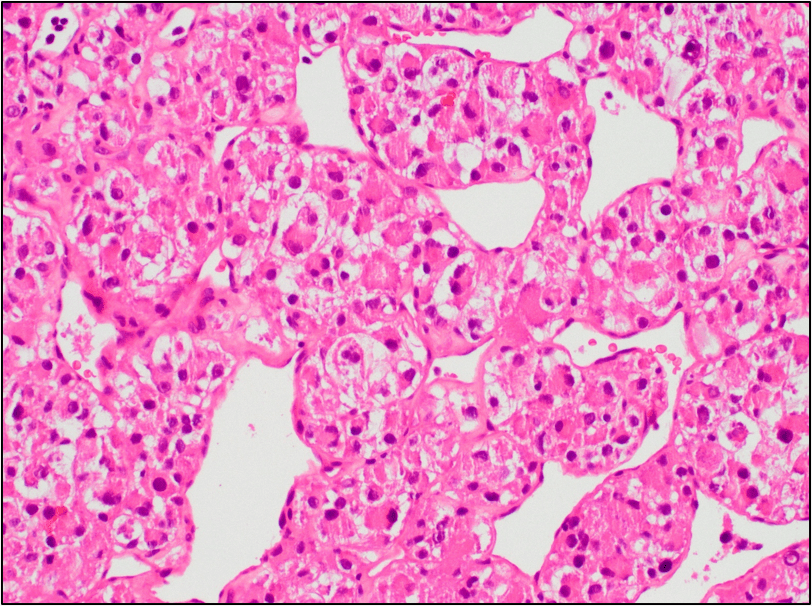
Figure 5. The tumor cells are large with distinct cell borders, ovoid nuclei, prominent nucleoli and abundant granular, eosinophilic and sometimes vacuolated cytoplasm (Hematoxylin and eosin, 40X).
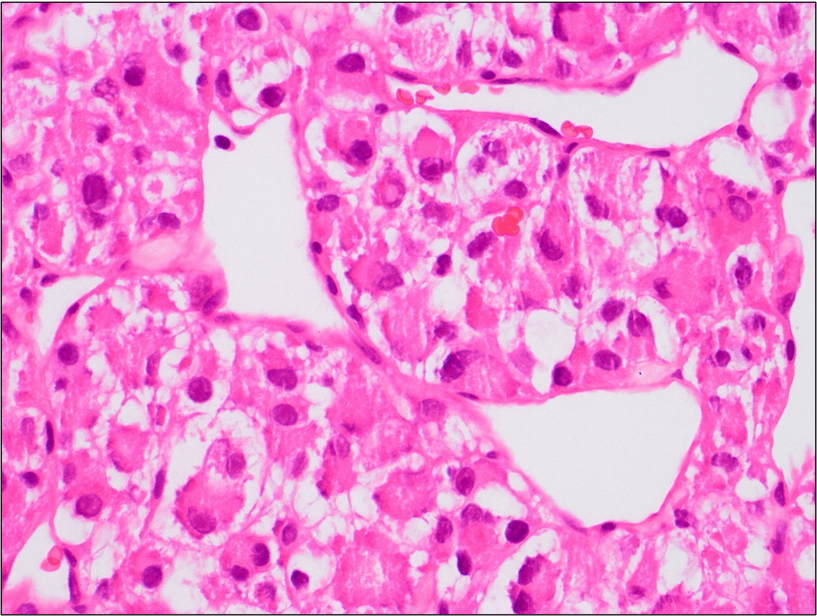
Figure 6. TFE3 Immunohistochemistry shows strong nuclear staining in the tumor cells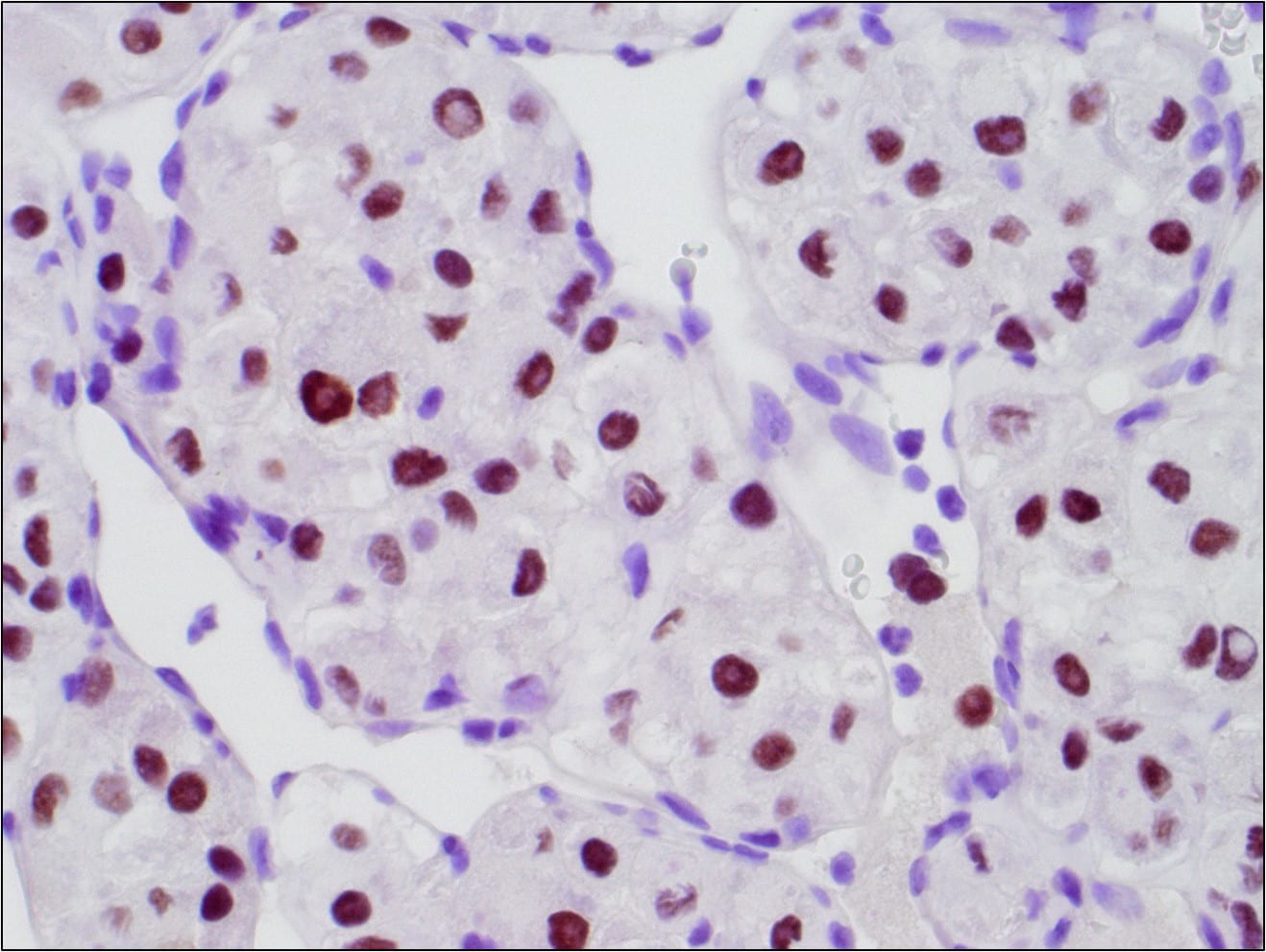
The tumor cells were immunohistochemically NEGATIVE for CK AE1/AE3, EMA, Vimentin, Glypican-3, Synaptophysin, SMA, Myo D1, S100, CD34, and PAX-8. Ki67 was positive in 20% of the cells.
Discussion
The final diagnosis for the touch preparation was malignant cells present (correlate with the concurrent surgical pathology case for further characterization). The final diagnosis for the 14G core needle biopsy was Alveolar soft part sarcoma.
Alveolar soft part sarcoma (ASPS) is a soft tissue sarcoma with distinct clinical and morphological features which accounts for approximately 0.4% to 1.0% of all soft tissue sarcomas
Clinical Features
ASPS usually occurs in adolescents and young adults and is most common in patients between the ages of 15 and 35. The tumor commonly occurs in different anatomic sites based on age. In infants and children the most common site is the head and neck region. In adults the most common site is the lower extremities. The tumor is slow growing and presents as a painless mass. The mass lesion usually does not cause functional impairment and can be overlooked resulting in many cases where metastasis to the lung and/or brain is the first manifestation of disease.
Pathologic Findings
Grossly ASPS tends to be a soft, friable and poorly circumscribed tumor. The cut surface is usually yellow-white to gray-red. Areas of necrosis and hemorrhage are common.
Microscopically the tumor is divided into compartments by thick fibrous trabeculae. The compartments are further subdivided into aggregates of tumor cells by sinusoidal vascular channels that are lined by endothelial cells. Necrosis and loss of cohesion between tumor cells is common. The tumor
cells are usually large, round to polygonal shaped with distinct cell borders, prominent irregular macronucleoli and abundant granular, eosinophilic and sometimes vacuolated cytoplasm. Mitotic figures are uncommon. Vascular invasion is a frequent finding.
PAS histochemical staining highlights intracellular glycogen and PAS-positive, diastase-resistant rhomboid or rod shaped crystals. Immunohistochemically the tumor cells are sometimes positive for MyoD1 and desmin (~50%).
The tumor cells are generally negative for keratins, EMA, neurofilaments, GFAP, HMB-45, melan-A and synaptophysin.
Cytogenetic and Molecular Findings
ASPS has a characteristic genetic alteration, der(17)t(X;17)(p11.2q25).
This alteration results in a fusion gene, ASPSCR1-TFE3, which encodes for a fusion protein that functions as an aberrant transcription factor leading to activation of MET signaling.
Prognosis
ASPS has a poor prognosis with an overall 5-year overall survival of around 20%. Metastatic disease usually occurs early with pulmonary, brain and skeleton metastases being the most common presentations.
However, metastatic disease may also occur years later in some cases. Older age at diagnosis, large tumor size, and the presence of metastatic disease at presentation are adverse prognostic parameters.
Treatment
Treatment of ASPS usually consists of radical resection of the primary tumor and metastatic lesions in addition to radiotherapy and/or chemotherapy.
References
1.Cibas, Edmund S., and Barbara S. Ducatman. Cytology: Diagnostic Principles and Clinical Correlates. Elsevier, 2021.
2.Goldblum, John, et al. Enzinger and Weiss’s Soft Tissue Tumors. 7th ed., Elsevier, 2019.